Технические характеристики Toyota RAV4 2022
Для входа в кабинет введите эл. почту или телефон и постоянный пароль и получите код авторизации.
К вашей учетной записи привязан номер телефона, не являющийся номером украинских операторов, отправка на этот номер невозможна
Вы можете воспользоваться функционалом по замене номера телефона
Для этого нажмите кнопку ниже
Не корректный адрес електронной почты
Телефон или эл. почта
Пароль
Код
Не получили код? Отправить код повторно
Напомнить пароль
Сменить номер телефона Обновление параметров входа
Фамилия *
Отчество
Эл. почта *
почта *
Телефон *
Пароль *
Код
Не получили код? Отправить код повторно
Ознакомлен с правилами пользования личным кабинетом
Уже зарегистрировался
Введите в поле эл. почту или номер телефона.Мы отправим вам смс c кодом для подтверждения учетной записи.
Телефон или эл. почта
почта
Код
Не получили код? Отправить код повторно
Новый пароль
Пароль должен быть не менее 6 символов, содержать цифры и латинские буквы, в том числе заглавные, и не должен совпадать с именем и эл. почтойВведите в поле свою эл. почту. Мы отправим вам письмо с кодом подтверждения на смену номера телефона
На указанный эл. почту был выслан код, введите его в поле для подтверждения вашей email.
почту был выслан код, введите его в поле для подтверждения вашей email.
Введите код который вы получили на новый номер телефон.
Технические характеристики Toyota Rav 4
Технические характеристики Тойота Рав 4. Узнайте габариты, расход топлива Toyota Rav 4 / Тойота Рав 4, особенности двигателей, подвесок, кузовов и прочих технических характеристик автомобилей Toyota Rav 4.
 png»/>
png»/>
Успей до 6 Ноября
1 479 380 ₽
1 972 500 ₽
Специальные условия на покупку Toyota Rav 4
Это обязательное поле
Наличие паспорта РФНажимая на кнопку выше вы даете согласие на обработку персональных данных
Технические характеристики
| Модификации | 2. 0 MT(149л.с.) 0 MT(149л.с.) | 2.0 CVT(149л.с.) | 2.0 CVT(149л.с.)4×4 | 2.5 AT(199л.с.)4×4 |
|---|---|---|---|---|
| Двигатель | ||||
| Количество передач | 6 | 8 | ||
| Марка топлива | Бензин с октановым числом 91 и выше | Бензин с октановым числом 91 и выше | Бензин с октановым числом 91 и выше | Бензин с октановым числом 91 и выше |
Мощность двигателя (л. с.) с.) | 149 | 149 | 149 | 199 |
| Объем двигателя | 2.0 | 2. 0 0 | 2.0 | 2.5 |
| Привод | Передний | Подключаемый полнопривод | Подключаемый полнопривод | Подключаемый полнопривод |
| Тип двигателя | Бензин с октановым числом 91 и выше | Бензин с октановым числом 91 и выше | Бензин с октановым числом 91 и выше | Бензин с октановым числом 91 и выше |
| Топливный бак (л) | 55 | 55 | 55 | 55 |
| Трансмиссия | MT | CVT | CVT | AT |
| Габаритные размеры | ||||
| Высота (мм) | 1685 | 1685 | 1685 | 1685 |
| Длина (мм) | 4600 | 4600 | 4600 | 4600 |
| Колесная база (мм) | 2690 | 2690 | 2690 | 2690 |
| Количество дверей | 5 | 5 | 5 | 5 |
| Количество мест | 5 | 5 | 5 | 5 |
| Объем багажника | 580 | 580 | 580 | 580 |
| Полная масса | 2015 | 2090 | 2170 | 2175 |
| Снаряженная масса | 1570-1575 | 1610-1620 | 1675-1730 | 1680-1740 |
| Ширина (мм) | 1855 | 1855 | 1855 | 1855 |
| Динамические характеристики | ||||
| Время разгона (0-100 км/ч, с) | 9,8 | 11 | 11 | 8,5 |
| Максимальная скорость (км/ч) | 190 | 190 | 190 | 200 |
| Подвеска | ||||
| Дорожный просвет (мм) | 195 | 195 | 195 | 195 |
| Задняя подвеска | независимая, пружинная | независимая, пружинная | независимая, пружинная | независимая, пружинная |
| Передняя подвеска | независимая, пружинная | независимая, пружинная | независимая, пружинная | независимая, пружинная |
| Размер колес | 225/65/R17 235/55/R19 225/60/R18 | |||
Основные характеристики
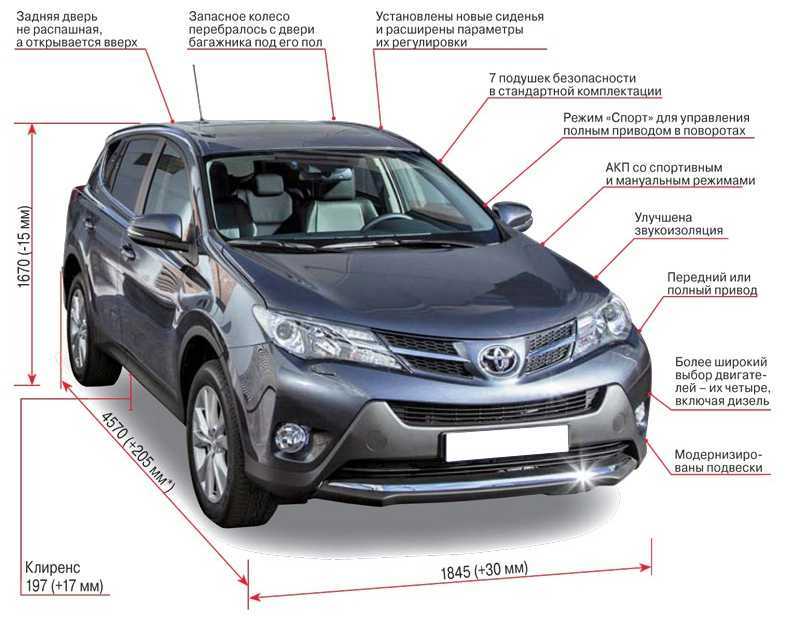
Длина кузова автомобиля достигает 4600 мм, ширина – 1855 мм, высота – 1685 мм. При этом колесная база составляет 2690 мм, клиренс – 195 мм.
При этом колесная база составляет 2690 мм, клиренс – 195 мм.
Он может оснащаться двигателем объемом 1,4; 1,6 л. Мощность силового агрегата достигает 100 л.с.; 123 л.с.
Машина может иметь механическую — МТ или автоматическую — АТ коробку переключения передач. МКПП понравится водителям, которые хотят полностью контролировать процесс езды. АКПП максимально удобна автолюбителям, которые не имеют большого стажа вождения.
До скорости 100 км/ч авто разгоняется за 9,8 с. Чтобы преодолеть расстояние 100 км при езде в смешанном режиме, понадобится от л горючего.
Полные технические характеристики Тойота Рав 4 2022 года выпуска Вы найдете на нашем сайте. Чтобы получить ответы на интересующие Вас вопросы, свяжитесь с менеджерами автосалона Селект Авто.
Экспресс заявка на кредит
Оставьте ваш номер и получите скидку, специальное предложение и подарок!
Это обязательное поле
Наличие паспорта РФ
Наличие паспорта РФ обязательно
Нажимая на кнопку выше вы даете согласие на обработку персональных данных
Заказать звонок
Мы перезвоним
через 30 секунд
Предпродажное уведомление 510(k) | FDA
Отправка электронных копий электронных копий (eCopy) или электронных шаблонов и ресурсов (eSTAR) на предварительные продажи медицинских устройств через Интернет
3 октября 2022 г. Портал сотрудничества с клиентами CDRH («Портал CDRH»).
Портал сотрудничества с клиентами CDRH («Портал CDRH»).
Основываясь на отслеживании прогресса подачи заявок 510(k), запущенном в 2021 г., и пробном процессе электронной загрузки, запущенном в июле 2022 г., портал CDRH теперь позволяет любому зарегистрировать учетную запись на портале CDRH для отправки предварительных заявок CDRH eCopy или eSTAR в режиме онлайн. .
Начиная с 1 октября 2023 все документы 510(k), кроме исключений*, должны подаваться в электронном виде с использованием eSTAR.
- Введение
- Что такое существенная эквивалентность
- Кто должен подавать форму 510(k)
- Когда требуется 510(k)
- Когда 510(k) не требуется
- Предварительные устройства
- Программа проверки третьей стороной
Обратите внимание: FDA взимает плату за рассмотрение предварительных уведомлений [510(k)]
Введение
Каждый человек, который хочет продавать в США устройства классов I, II и III, предназначенные для использования человеком, для которых не требуется предварительная регистрация (PMA), должен подать заявку 510(k) FDA, если только устройство не освобождено от требований 510(k) Федерального закона о пищевых продуктах, лекарствах и косметических средствах (Закон FD&C) и не превышает ограничения исключений в . 9 глав правил классификации устройств (например, 21 CFR). 862.9, 21 CFR 864.9). Формы 510(k) нет; тем не менее, 21 CFR 807 Subpart E описывает требования к представлению 510 (k). Прежде чем продавать устройство, каждый заявитель должен получить заказ в форме письма от FDA, в котором устройство будет признано практически эквивалентным (SE) и указано, что устройство может продаваться в США. Этот приказ «очищает» устройство. для коммерческого распространения (см. Руководство по программе 510(k)).
9 глав правил классификации устройств (например, 21 CFR). 862.9, 21 CFR 864.9). Формы 510(k) нет; тем не менее, 21 CFR 807 Subpart E описывает требования к представлению 510 (k). Прежде чем продавать устройство, каждый заявитель должен получить заказ в форме письма от FDA, в котором устройство будет признано практически эквивалентным (SE) и указано, что устройство может продаваться в США. Этот приказ «очищает» устройство. для коммерческого распространения (см. Руководство по программе 510(k)).
A 510(k) — это предварительная заявка, подаваемая в FDA для демонстрации того, что продаваемое устройство является таким же безопасным и эффективным, то есть по существу эквивалентно легально продаваемому устройству (раздел 513(i)(1)(A). ) Закон о FD&C). Заявители должны сравнить свое устройство с одним или несколькими аналогичными устройствами, продаваемыми на законных основаниях, а также сделать и подтвердить свои существенные заявления об эквивалентности. Легально продаваемое устройство — это устройство, которое продавалось на законных основаниях до 28 мая 1976 г. (устройство с предварительными поправками), или устройство, которое было реклассифицировано из класса III в класс II или I, устройство, которое было признано SE через 510(k ) или устройство, получившее регистрационное удостоверение в процессе классификации De Novo в соответствии с разделом 513(f)(2) Закона FD&C, на которое не распространяются требования о предварительном уведомлении. Легально продаваемое устройство (устройства), к которому применяется эквивалентность, широко известно как «предикат». Хотя устройства, недавно прошедшие очистку в соответствии со статьей 510(k), часто выбираются в качестве предиката, эквивалентность которого заявлена, любое легально продаваемое устройство может использоваться в качестве предиката. . Легальный маркетинг также означает, что предикат не может быть предикатом, нарушающим Закон FD&C.
(устройство с предварительными поправками), или устройство, которое было реклассифицировано из класса III в класс II или I, устройство, которое было признано SE через 510(k ) или устройство, получившее регистрационное удостоверение в процессе классификации De Novo в соответствии с разделом 513(f)(2) Закона FD&C, на которое не распространяются требования о предварительном уведомлении. Легально продаваемое устройство (устройства), к которому применяется эквивалентность, широко известно как «предикат». Хотя устройства, недавно прошедшие очистку в соответствии со статьей 510(k), часто выбираются в качестве предиката, эквивалентность которого заявлена, любое легально продаваемое устройство может использоваться в качестве предиката. . Легальный маркетинг также означает, что предикат не может быть предикатом, нарушающим Закон FD&C.
До тех пор, пока отправитель не получит заказ на объявление SE устройства, он не может приступить к продаже устройства. Как только устройство будет определено как SE, оно может быть продано в США.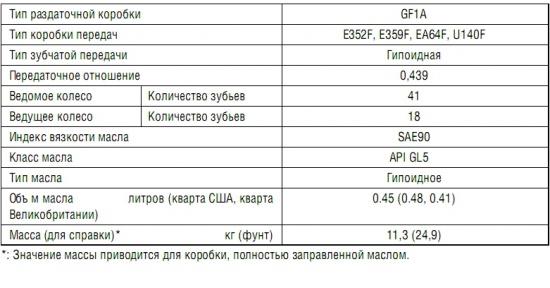 Определение SE обычно делается в течение 90 дней и делается на основе информации, предоставленной заявителем.
Определение SE обычно делается в течение 90 дней и делается на основе информации, предоставленной заявителем.
Обратите внимание, что FDA обычно не проводит инспекции 510(k) на предприятиях перед таможенной очисткой. Заявитель может продавать устройство сразу после получения разрешения 510(k). Производитель должен быть готов к проверке системы качества FDA (21 CFR 820) в любое время после получения разрешения 510(k).
Что такое существенная эквивалентность
A 510(k) требует демонстрации существенной эквивалентности другому устройству, легально продаваемому в США. Существенная эквивалентность означает, что новое устройство так же безопасно и эффективно, как предикат.
Средство по существу эквивалентно, если по сравнению с предикатом оно:
- имеет такое же предназначение, как и предикат; и
- имеет те же технологические характеристики, что и предикат;
или - имеет то же предназначение, что и предикат; и
- имеет другие технологические характеристики и не вызывает других вопросов безопасности и эффективности; и
- информация, представленная FDA, демонстрирует, что устройство так же безопасно и эффективно, как и легально продаваемое устройство.

Заявление о существенной эквивалентности не означает, что новые и предикатные устройства должны быть идентичными. FDA сначала устанавливает, что новые и стандартные устройства имеют одинаковое предполагаемое использование, и любые различия в технологических характеристиках не вызывают разных вопросов безопасности и эффективности. Затем FDA определяет, является ли устройство таким же безопасным и эффективным, как и предикатное устройство, анализируя научные методы, используемые для оценки различий в технологических характеристиках и рабочих характеристиках. Эти данные о производительности могут включать клинические данные и неклинические данные о производительности, включая испытания инженерных характеристик, стерильность, электромагнитную совместимость, проверку программного обеспечения, оценку биосовместимости и другие данные.
Устройство не может продаваться в США до тех пор, пока заявитель не получит письмо о том, что устройство по существу эквивалентно. Если FDA определяет, что устройство не является по существу эквивалентным , заявитель может:
- повторно подать другую заявку 510(k) с новыми данными,
- запросить класс I или II в процессе классификации De Novo
- подать заявление о реклассификации или
- подать заявку на допродажное утверждение (PMA).

Кто должен подавать форму 510(k)
Закон FD&C и положение 510(k) (21 CFR 807) не определяют, кто должен подавать форму 510(k). Вместо этого они указывают, какие действия, такие как вывод устройства на рынок США, требуют представления 510 (k).
Следующие четыре категории сторон должны подать форму 510(k) в FDA:
- Отечественные производители, выводящие устройство на рынок США;
Производители готовых устройств должны подать форму 510(k), если они производят устройство в соответствии со своими спецификациями и продают его в США. Аксессуары к готовым устройствам, продаваемые конечному пользователю, также считаются готовыми устройствами. Однако производители компонентов устройства не обязаны подавать форму 510(k), если только такие компоненты не рекламируются для продажи конечному пользователю в качестве запасных частей. Контрактные производители, те фирмы, которые производят устройства по контракту в соответствии с чужими спецификациями, не обязаны подавать форму 510(k).

- Разработчики спецификации, выводящие устройство на рынок США;
Разработчик спецификаций разрабатывает спецификации для готового устройства, но производит устройство по контракту с другой фирмой или организацией. Разработчик спецификации представляет 510(k), а не контрактный производитель.
- Переупаковщики или перемаркировщики, которые вносят изменения в маркировку или чьи операции существенно влияют на устройство.
От переупаковщиков или лиц, переклеивающих этикетку, может потребоваться представить форму 510(k), если они существенно изменят маркировку или иным образом повлияют на какое-либо состояние устройства. Существенные изменения маркировки могут включать модификацию руководств, например добавление нового предполагаемого использования, удаление или добавление предупреждений, противопоказаний и т. д. Такие операции, как стерилизация, могут изменить состояние устройства. Однако большинство компаний, занимающихся переупаковкой или перемаркировкой, не обязаны подавать форму 510(k).

- Иностранные производители/экспортеры или представители иностранных производителей/экспортеров в США, представляющие устройство на рынке США.
Обратите внимание, что все производители (включая разработчиков спецификаций) устройств классов II и III и некоторых устройств класса I обязаны следовать правилам проектирования (21 CFR 820.30) при разработке своих устройств. Владелец формы 510(k) должен иметь документацию по контролю конструкции, доступную для рассмотрения FDA во время инспекции объекта. Кроме того, любые изменения в спецификациях устройства или производственных процессах должны производиться в соответствии с положением о системе качества (21 CFR 820) и могут подпадать под действие нового раздела 510(k). Дополнительную информацию можно найти на веб-странице «Нужен ли новый 510(k) для модификации устройства?»
Когда требуется форма 510(k)
Форма 510(k) требуется, когда:
- Если не указано иное, ввод устройства в коммерческое распространение (маркетинг) в первый раз.
 После 28 мая 1976 г. (дата вступления в силу поправок к Закону о медицинских устройствах) любой, кто хочет продать устройство в США, должен подать заявку 510 (k) не менее чем за 90 дней до выставления устройства на продажу. даже если он находился в стадии разработки или клинических исследований до этой даты. Если ваше устройство не было продано вашей фирмой до 28 мая 19 г.76, требуется 510(k).
После 28 мая 1976 г. (дата вступления в силу поправок к Закону о медицинских устройствах) любой, кто хочет продать устройство в США, должен подать заявку 510 (k) не менее чем за 90 дней до выставления устройства на продажу. даже если он находился в стадии разработки или клинических исследований до этой даты. Если ваше устройство не было продано вашей фирмой до 28 мая 19 г.76, требуется 510(k). - Произошло изменение или модификация легально продаваемого устройства, и это изменение может существенно повлиять на его безопасность или эффективность. Бремя решения о том, может ли модификация существенно повлиять на безопасность или эффективность устройства, лежит на держателе 510(k). Любые модификации должны быть сделаны в соответствии с положением о системе качества, 21 CFR 820, и зарегистрированы в основной записи устройства и записях контроля изменений. Рекомендуется записывать обоснование подачи или непредставления нового 510(k) в записях контроля изменений.
Требуется новая заявка 510(k) для внесения изменений или модификаций в существующее устройство, если модификации могут существенно повлиять на безопасность или эффективность устройства или если устройство должно продаваться для нового или другого предполагаемого использования.
 См. Требуется ли новый 510(k) для модификации устройства? для получения дополнительной информации.
См. Требуется ли новый 510(k) для модификации устройства? для получения дополнительной информации.
Когда 510(k) не требуется
Ниже приведены примеры случаев, когда 510(k) не требуется.
- Вы продаете незавершенные устройства другой фирме для дальнейшей обработки или продаете компоненты, которые будут использоваться при сборке устройств другими фирмами. Однако, если ваши компоненты будут продаваться непосредственно конечным пользователям в качестве запасных частей, потребуется 510(k).
- Ваше устройство не продается и не распространяется в коммерческих целях. Вам не нужен 510(k) для разработки, оценки или тестирования устройства. Это включает клиническую оценку. Обратите внимание, что если вы проводите клинические испытания с вашим устройством, вы подпадаете под действие положения об исключении для исследовательских устройств (IDE) (21 CFR 812).
- Вы распространяете устройство отечественного производства другой фирмы. Вы можете разместить на устройстве этикетку «Распространяется фирмой ABC» или «Изготовлено для фирмы ABC» (21 CFR 801.
 1) и продавать его конечным пользователям без подачи 510(k).
1) и продавать его конечным пользователям без подачи 510(k). - В большинстве случаев, если вы занимаетесь переупаковкой или перемаркировкой, и существующая маркировка или состояние устройства существенно не изменились. Маркировка должна соответствовать маркировке, представленной в 510(k), с теми же показаниями к применению, предупреждениями и противопоказаниями.
- Ваше устройство легально продавалось в коммерческих целях до 28 мая 1976 г. и не претерпело значительных изменений или модификаций по конструкции, компонентам, способу изготовления или предполагаемому использованию. Эти устройства «дедушкины», и у вас есть документация о статусе предварительной поправки, подтверждающая это.
- Устройство произведено за пределами США, и вы являетесь импортером медицинского устройства иностранного производства. Форма 510(k) не требуется, если 510(k) был представлен иностранным производителем и получил разрешение на продажу. Как только иностранный производитель получил разрешение 510(k) на устройство, иностранный производитель может экспортировать свое устройство любому импортеру в США.

- На ваше устройство не распространяется действие 510(k) постановления (21 CFR 862-892). То есть некоторые устройства Класса I или II могут впервые продаваться без подачи 510(k). Список освобожденных устройств класса I и II можно найти в Исключениях для медицинских устройств 510 (k) и Требованиях GMP. Однако, если устройство превышает ограничения исключений в .9глав правил классификации устройств (например, 21 CFR 862.9, 21 CFR 864.9), например, устройство имеет другое предназначение или работает с использованием другой фундаментальной научной технологии, чем законно продаваемое устройство в этом родовом типе устройства, или устройство является переработанным одноразовым устройством, то для продажи нового устройства необходимо подать форму 510(k).
Устройства с предварительной поправкой
Термин «устройство с предварительной поправкой» относится к устройствам, законно проданным фирмой в США до 28 мая 19 года.76 и , которые с тех пор не претерпели:
- существенных изменений или модификаций; и
- , для которого FDA не опубликовало положение, требующее подачи заявки на PMA.

Устройства, отвечающие вышеуказанным критериям, являются «устаревшими» устройствами и не требуют 510(k). Устройство должно иметь такое же предназначение, как и предназначенное для продажи до 28 мая 1976 г. Если устройство маркировано для другого предполагаемого использования, то устройство считается новым устройством, и 510 (k) должно быть представлено в FDA для разрешения на продажу. .
Обратите внимание, что вы должны быть владельцем устройства на рынке до 28 мая 1976 года, чтобы устройство стало устаревшим. Если ваше устройство похоже на устаревшее устройство и поступило в продажу под номером после 28 мая 1976 года, то ваше устройство НЕ соответствует требованиям, предъявляемым к устаревшему, и вы должны подать форму 510(k). Чтобы фирма могла заявить, что у нее есть устройство для предварительных поправок, она должна продемонстрировать, что это устройство было маркировано, рекламировано и распространялось в межгосударственной торговле для конкретного предполагаемого использования и что предполагаемое использование не изменилось.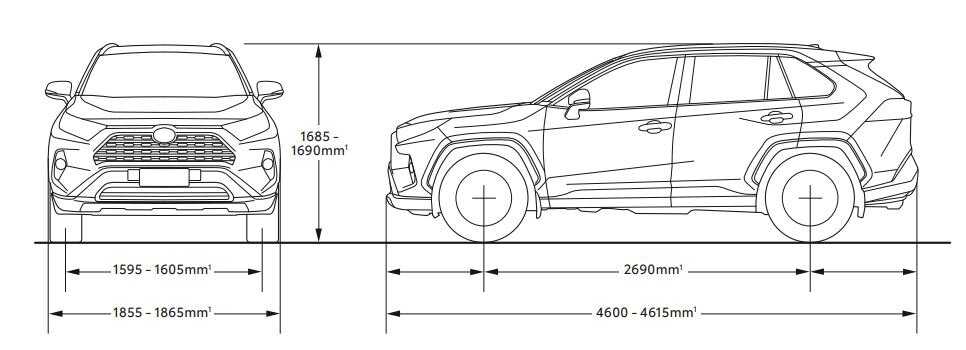 См. Статус предварительной поправки для получения информации о требованиях к документации.
См. Статус предварительной поправки для получения информации о требованиях к документации.
Программа проверки третьей стороной
Центр устройств и радиологического здоровья (CDRH) внедрил Программу проверки третьей стороной. Эта программа предоставляет производителям определенных устройств возможность отправлять свои 510(k) частным сторонам (признанным третьим сторонам), определенным FDA, для рассмотрения, вместо того, чтобы отправлять их напрямую в CDRH. Для получения дополнительной информации о программе, соответствующих устройствах и списке признанных третьих лиц перейдите на страницу информации о программе проверки сторонних поставщиков.
Дополнительная информация
- Отправка и отслеживание предпродажных заявок на медицинские изделия онлайн: портал CDRH
- Модуль обучения CDRH: программа 510(k)
- 510(k) Часто задаваемые вопросы
- Процесс классификации De Novo (оценка автоматического обозначения класса III) — Руководство для промышленности и персонала Управления по санитарному надзору за качеством пищевых продуктов и медикаментов
- 510(k) Блок-схема принятия решений
- Программа 510(k): Оценка существенной эквивалентности в предпродажных уведомлениях [510(k)] — Руководство для промышленности и персонала Управления по контролю за продуктами и лекарствами
- Принятие решения о том, когда подавать 510(k) на изменение существующего устройства
- Принятие решения о том, когда подавать 510(k) на изменение программного обеспечения существующего устройства
- 510(k) Зазоры
Связаться с FDA
1 (800) 638-2041
(301) 796-7100
DICE@fda. hhs.gov
hhs.gov
Информационно-медицинские устройства / радиационные продукты
Отдел промышленности и обучения потребителей
Центр обучения CDRH и радиологическое здоровье
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов
10903 New Hampshire Avenue
Silver Spring, MD 20993
Компьютерное моделирование и модели рабочих характеристик спектрально эквивалентных рентгеновских лучей в медицинской диагностической радиологии
. 2007 г., 32 октября (4): 175–84.
дои: 10.4103/0971-6203.37483.
Акинтунде А Окунаде 1
принадлежность
- 1 Факультет физики Университета Обафеми Аволово 220005, Иле-Ифе Осун, Нигерия.
- PMID: 21224928
- PMCID: PMC3014103
- DOI:
10.
 4103/0971-6203.37483
4103/0971-6203.37483
Бесплатная статья ЧВК
Акинтунде А Окунаде. J Med Phys. 2007 Октябрь
Бесплатная статья ЧВК
. 2007 г., 32 октября (4): 175–84.
дои: 10.4103/0971-6203.37483.
Автор
Акинтунде А Окунаде 1
принадлежность
- 1 Факультет физики, Университет Обафеми Аволово 220005, Иле-Ифе Осун, Нигерия.
- PMID: 21224928
- PMCID: PMC3014103
- DOI:
10.
 4103/0971-6203.37483
4103/0971-6203.37483
Абстрактный
Для достижения единообразия рентгенологической визуализации рекомендуется использовать понятие эквивалентности по форме (качеству) и размеру (количеству) пучков клинического рентгеновского излучения для проведения сравнительной оценки изображения и дозы облучения пациента. При использовании в одной и той же геометрии облучения рентгеновские лучи, строго или относительно эквивалентные по форме и размеру, будут давать одинаковое или относительно одинаковое качество изображения и дозу облучения пациента. Простые математические модели и программа EQSPECT.FOR были разработаны для сравнительной оценки рабочих характеристик по контрасту (C), отношению контраста к шуму (CNR) и добротности (FOM = CNR(2)/DOSE). для спектрально эквивалентных лучей, прошедших через фильтрующие материалы, называются обычными и k-краевыми. При одном и том же значении рабочего потенциала (кВп) результаты показывают, что спектрально эквивалентный пучок, прошедший через обычный фильтр с более высоким атомным номером (значением Z) по сравнению с прошедшим через обычный фильтр с более низким значением Z, дал такое же значение C и ФОМ. Однако по сравнению со спектрально эквивалентным лучом, прошедшим через фильтр с более низким значением Z, пучок через фильтр с более высоким значением Z дает более высокие значения CNR и DOSE при одинаковой нагрузке трубки (мАс) и кВп. При условии эквивалентности спектра, при масштабированной (или уменьшенной) загрузке трубки и одинаковом kVp, фильтрующие материалы с более высоким значением Z могут давать те же значения C, CNR, DOSE и FOM, что и фильтрующие материалы с более низким значением Z. В отличие от случая сравнения спектрально эквивалентного луча, прошедшего через один обычный фильтр, и луча, прошедшего через другой обычный фильтр, невозможно вывести простые математические формулировки относительной производительности спектрально эквивалентного луча, прошедшего через данный обычный фильтрующий материал, и через фильтр Кеджа. материал.
Однако по сравнению со спектрально эквивалентным лучом, прошедшим через фильтр с более низким значением Z, пучок через фильтр с более высоким значением Z дает более высокие значения CNR и DOSE при одинаковой нагрузке трубки (мАс) и кВп. При условии эквивалентности спектра, при масштабированной (или уменьшенной) загрузке трубки и одинаковом kVp, фильтрующие материалы с более высоким значением Z могут давать те же значения C, CNR, DOSE и FOM, что и фильтрующие материалы с более низким значением Z. В отличие от случая сравнения спектрально эквивалентного луча, прошедшего через один обычный фильтр, и луча, прошедшего через другой обычный фильтр, невозможно вывести простые математические формулировки относительной производительности спектрально эквивалентного луча, прошедшего через данный обычный фильтрующий материал, и через фильтр Кеджа. материал.
Ключевые слова: контраст; элементарные фильтры; медицинская диагностическая радиология.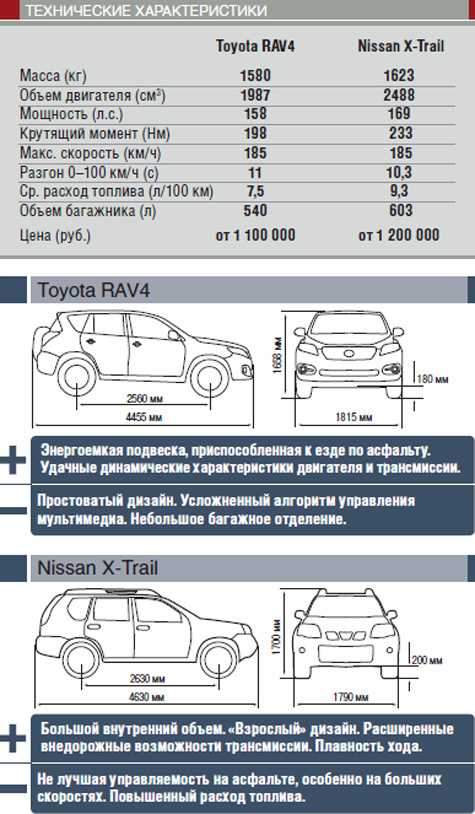
Заявление о конфликте интересов
Конфликт интересов: не объявлено.
Цифры
Рисунок 1
Принципиальная схема реализации…
Рисунок 1
Принципиальная схема для реализации компьютерного моделирования
фигура 1Принципиальная схема для реализации компьютерного моделирования
Рисунок 2
Отношения T, DOSE, [CNR]…
Рисунок 2
Отношения T, DOSE, [CNR] 2 , CNR, контраст и FOM для лучей…
фигура 2 Отношения T, DOSE, [CNR] 2 , CNR, контрастность и FOM для лучей, прошедших через алюминиево-медный фильтр (знаменатель) и выбранные альтернативные фильтрующие материалы (числитель) при «спектрально» эквивалентной толщине. Это для усиливающего экрана 80 мг/см −2 Gd 2 O 2 S, контрастного вещества 10 мг/см −2 йод и объект водяного фантома толщиной 20 см. Значения среднеквадратичной ошибки менее 1,0% для всех случаев согласования упрочнения пар обычных фильтров
Это для усиливающего экрана 80 мг/см −2 Gd 2 O 2 S, контрастного вещества 10 мг/см −2 йод и объект водяного фантома толщиной 20 см. Значения среднеквадратичной ошибки менее 1,0% для всех случаев согласования упрочнения пар обычных фильтров
Рисунок 3
Сравнение формы и…
Рисунок 3
Сравнение формы и размера передаваемого спектрального распределения энергии при «спектральном»…
Рисунок 3Сравнение формы и размера пропускаемого спектрального распределения энергии при «спектрально» эквивалентной толщине алюминиевого и медного фильтрующих материалов. Сравниваются спектры (i) без масштабирования, Φ Al (E i ) и Φ Cu (E i ) и (ii) с масштабированием, Φ Al (E i ) и aΦ Cu (Е и )
Рисунок 4
Сравнение формы и…
Рисунок 4
Сравнение формы и размера передаваемого спектрального распределения энергии при «спектральном»…
Рисунок 4 Сравнение формы и размера пропускаемого спектрального распределения энергии при «спектрально» эквивалентной толщине алюминиево-медных и гадолиниевых фильтров. Значения минимальной среднеквадратичной ошибки согласования упрочнения оказались равными 24,5 % для этих алюминиевых и гадолиниевых фильтров и 44,4 % для этих медных и гадолиниевых фильтров. Гадолиниевый фильтр пропускает больше фотонов с энергиями ниже k-края, чем алюминиевые и медные фильтры. Существует значительное различие в упрочняющих и затухающих свойствах алюминия/меди (обычный фильтр) и гадолиния (фильтр с k-краем)
Значения минимальной среднеквадратичной ошибки согласования упрочнения оказались равными 24,5 % для этих алюминиевых и гадолиниевых фильтров и 44,4 % для этих медных и гадолиниевых фильтров. Гадолиниевый фильтр пропускает больше фотонов с энергиями ниже k-края, чем алюминиевые и медные фильтры. Существует значительное различие в упрочняющих и затухающих свойствах алюминия/меди (обычный фильтр) и гадолиния (фильтр с k-краем)
Рисунок 5
Отношения T, DOSE, [CNR]…
Рисунок 5
Отношения T, DOSE, [CNR] 2 , CNR, контраст и FOM для лучей…
Рисунок 5 Отношения T, DOSE, [CNR] 2 , CNR, контраст и FOM для лучей, прошедших через алюминиево-медный фильтр (знаменатель) и лантаниевый фильтр (числитель) при «спектрально» эквивалентных толщинах. Это для усиливающего экрана 80 мг/см −2 Gd 2 O 2 S, контрастное вещество 10 мг/см −2 йод и объект водяного фантома толщиной 20 см. Существует значительная разница как в форме, так и в размере пучков, пропускаемых алюминиево-медными и лантаниевыми фильтрами. Следовательно, отношения T, DOSE, [CNR] 2 , CNR, контраст и FOM значительно отклоняются от единицы
Это для усиливающего экрана 80 мг/см −2 Gd 2 O 2 S, контрастное вещество 10 мг/см −2 йод и объект водяного фантома толщиной 20 см. Существует значительная разница как в форме, так и в размере пучков, пропускаемых алюминиево-медными и лантаниевыми фильтрами. Следовательно, отношения T, DOSE, [CNR] 2 , CNR, контраст и FOM значительно отклоняются от единицы
См. это изображение и информацию об авторских правах в PMC
Похожие статьи
Параметризованные алгоритмы количественной дифференциации спектрально-эквивалентных медицинских диагностических рентгеновских лучей.
Окунаде А.А. Окунаде АА. мед. физ. 2005 июнь; 32 (6): 1785-95. дои: 10.1118/1.1915959. мед. физ. 2005. PMID: 16013736
Сравнение качества изображения между протоколами КТ с одной и двумя энергиями для визуализации печени.
Yao Y, Ng JM, Megibow AJ, Pelc NJ. Яо Ю и др. мед. физ. 2016 авг; 43 (8): 4877. дои: 10.1118/1.4959554. мед. физ. 2016. PMID: 27487905
Оптимизация дозы при рентгенографии сердца у детей.
Гисласон А.Дж., Дэвис А.Г., Коуэн А.Р. Гисласон А.Дж. и соавт. мед. физ. 2010 окт; 37 (10): 5258-69. дои: 10.1118/1.3488911. мед. физ. 2010. PMID: 21089760
Имитационное исследование квазимонохроматического луча для рентгеновской компьютерной маммотографии.
McKinley RL, Tornai MP, Samei E, Bradshaw ML. МакКинли Р.Л. и др. мед. физ. 2004 г., 31 апреля (4): 800-13. дои: 10.1118/1.1668371. мед. физ. 2004. PMID: 15124997
Спектральная КТ со счетом фотонов: улучшенное разложение материала с помощью рентгеновских лучей, отфильтрованных по K-краю.

Шихалиев П.М. Шихалиев ПМ. физ.-мед. биол. 2012 21 марта; 57 (6): 1595-615. дои: 10.1088/0031-9155/57/6/1595. Epub 2012 7 марта. физ.-мед. биол. 2012. PMID: 22398007
Посмотреть все похожие статьи
использованная литература
- Бун Дж. М. Эквивалентные спектры как показатель качества пучка. мед. физ. 1986; 13: 861–8. — пабмед
- Бун Дж. М. Эквивалентные спектры с тремя параметрами как показатель качества пучка. мед. физ. 1988; 15: 304–10. — пабмед
- Тореус Р.

- Тореус Р.