Пандемия в роли катализатора: COVID-19 стал стимулом к решению приоритетных задач в области психического здоровья
9 октября 2021
Пандемия COVID-19, несомненно, вызвала кардинальные перемены как в повседневной жизни каждого из нас, так и нашего глобального общества в целом. Поодиночке и сообща мы пытались понять, что представляет собой этот вирус, перевернувший наш мир; разобраться в том изобилии информации – и дезинформации, – которая служила руководством к нашим ответным мерам; оценить риск для наших семей и сообществ; и разработать стратегии, позволяющие адаптироваться к новым требованиям, перебороть возникшие страхи и разрешить стоящие пред нами дилеммы.
Оптимистично настроенная часть нашего общества надеется, что худшие дни пандемии уже позади. В Соединенных Штатах большинство детей снова ходят в школу, многие из нас вернулись на свои рабочие места, и жизнь, по крайней мере частично, входит в привычный ритм, присущий ей до 2020 года. Однако COVID-19, который более полутора лет преобладал во всех аспектах нашей жизни, усугубляет еще одну, не менее опасную пандемию, связанную с ростом психических заболеваний.
Однако COVID-19, который более полутора лет преобладал во всех аспектах нашей жизни, усугубляет еще одну, не менее опасную пандемию, связанную с ростом психических заболеваний.
Проблемы психического здоровья не новы, однако пандемия COVID-19 быстро спровоцировала углубление кризиса. В 2019 году, до начала пандемии, каждый пятый взрослый житель Соединенных Штатов страдал каким-либо психическим заболеванием – чаще всего депрессией или тревожным расстройством; кроме того, ожидалось, что с проблемой психического здоровья в течение своей жизни столкнутся половина американцев1. Сегодня эти цифры значительно выше, и некоторые исследования показывают, что до 80 процентов американцев испытывают тревожные чувства, депрессию, уныние или одиночество.
Это неудивительно, учитывая, что COVID-19 продолжает сеять хаос, оставляя все возрастающий след опустошения и горя по всему миру, по мере того как мы привыкаем к «новой норме». С декабря 2019 года по всему миру от COVID-19 скончались свыше 4,8 миллиона человек, зарегистрировано более 236 миллионов случаев заболевания.
Последствия этого экстремального кризиса в области общественного здравоохранения в сочетании с сопутствующими экономическими и социальными потрясениями будут сказываться на протяжении десятилетий, а то и на протяжении жизни целого поколения. Как мать и психолог, я не могу не задумываться о воздействии этой пандемии на детей во всем мире. Для большинства детей школьная жизнь сменилась дистанционным обучением на дому, лишенным социально-эмоционального компонента, который имеет решающее значение для их здорового развития. Мы еще долгое время будем переживать потери, причиненные пандемией, будь то работа, бизнес, средства к существованию, столь любимое нами время общения с близкими, и более всего – безвременно покинувших нас дорогих сердцу членов семей и сообществ.

Тем не менее пандемия COVID-19 не только привела к росту проблем с психическим здоровьем; она также послужила стимулом к активизации стремления наших сообществ к повышению осведомленности об этих проблемах и к расширению доступа к поддержке и услугам для тех, кого они затрагивают.
Прежде всего следствием широкомасштабного воздействия COVID-19 стало то, что диалог о психическом здоровье приобрел более открытый характер, а наши знания в этой области теперь глубже, чем когда-либо прежде. Очень многие из нас знают кого-то, кто в результате пандемии испытывает повышенный стресс, тревогу или другие психологические проблемы, что заставляет нас искать поддержку у своих семей, друзей и коллег, а также обращаться за помощью к услугам специалистов в области психического здоровья.
Пандемия стала катализатором усилий, которые уже давно предпринимают профессиональные сообщества специалистов в области психического и физического здоровья, с тем чтобы обеспечить дестигматизацию проблем психического здоровья, а обращение за помощью при наличии таких проблем сделать вполне естественным. Психическое здоровье является неотъемлемой составляющей здоровья человека, и мы должны в равной степени заниматься вопросами физического и психического здоровья. Мы видим, что в самых разных сообществах и отраслях более открыто обсуждаются самочувствие людей, проблемы, с которыми они сталкиваются, и виды ресурсов, которые они используют для решения этих проблем. Общественное восприятие психических заболеваний движется в сторону смягчения, а тема эта обсуждается чаще, более открыто и в более инклюзивном порядке, особенно среди молодежи.
Психическое здоровье является неотъемлемой составляющей здоровья человека, и мы должны в равной степени заниматься вопросами физического и психического здоровья. Мы видим, что в самых разных сообществах и отраслях более открыто обсуждаются самочувствие людей, проблемы, с которыми они сталкиваются, и виды ресурсов, которые они используют для решения этих проблем. Общественное восприятие психических заболеваний движется в сторону смягчения, а тема эта обсуждается чаще, более открыто и в более инклюзивном порядке, особенно среди молодежи.
Д-р Мишель Ниэлон, президент Чикагской школы профессиональной психологии.
Кроме того, в условиях пандемии COVID-19 стало невозможным игнорировать расовое неравенство в плане того, как люди с иным цветом кожи переживают проблемы психического здоровья и в какой мере они могут воспользоваться соответствующими услугами. Нынешний кризис со всей очевидностью продемонстрировал, что в западном обществе люди с иным цветом кожи имеют меньший доступ к медицинской и психологической помощи, а вероятность их обращения за такой помощью ниже. Мы как общество обязаны пересмотреть структуру вариантов оказания соответствующей помощи в целях обеспечения всеохватного и равного доступа для всех сообществ. В целом нам требуется больше не просто психотерапевтов и поставщиков услуг в области психического здоровья, но соответствующих специалистов из разных слоев общества, которые могут лучше сопереживать людям с подобным опытом и поддерживать их.
Мы как общество обязаны пересмотреть структуру вариантов оказания соответствующей помощи в целях обеспечения всеохватного и равного доступа для всех сообществ. В целом нам требуется больше не просто психотерапевтов и поставщиков услуг в области психического здоровья, но соответствующих специалистов из разных слоев общества, которые могут лучше сопереживать людям с подобным опытом и поддерживать их.
Мы все должны сыграть свою роль в содействии этим изменениям в нашей системе здравоохранения. Как президент крупного университета, специализирующегося на вопросах психологии, науки о поведенческом здоровье и сестринском деле, я неустанно уделяю внимание набору и выпуску учащихся, которые отражают многообразную идентичность своих сообществ, что позволяет непрерывно пополнять контингент самых разных специалистов, способных коренным образом улучшить здоровье и благополучие будущих поколений.
Пандемия COVID-19 также резко ускорила рост возможностей телемедицины, что помогает расширить доступ к услугам. В начале 2020 года вся наша отрасль с впечатляющей скоростью перешла от очного предоставления медицинских услуг к онлайновому. Это испытание на прочность «здесь и сейчас» подтолкнуло внедрение по нарастающей инноваций в виртуальной терапии: мы увидели успешное взаимодействие между пациентами и поставщиками услуг, повышение качества доступных платформ и услуг, а также рост инвестиций в новые технологии. И, хотя многие возвращаются к личному общению и очным вариантам предоставления медицинских услуг, телемедицина будет оставаться важным средством расширения доступа к новым группам людей, ищущих помощи.
В начале 2020 года вся наша отрасль с впечатляющей скоростью перешла от очного предоставления медицинских услуг к онлайновому. Это испытание на прочность «здесь и сейчас» подтолкнуло внедрение по нарастающей инноваций в виртуальной терапии: мы увидели успешное взаимодействие между пациентами и поставщиками услуг, повышение качества доступных платформ и услуг, а также рост инвестиций в новые технологии. И, хотя многие возвращаются к личному общению и очным вариантам предоставления медицинских услуг, телемедицина будет оставаться важным средством расширения доступа к новым группам людей, ищущих помощи.
Наконец, на фоне нашего продолжающегося выхода из самого тяжелого этапа пандемии и возвращения к работе мы наблюдаем значительный сдвиг в том, как обсуждение вопросов психического здоровья интегрируется в нашу культуру труда. Вызванный COVID-19 кризис заставил многих работодателей взять на себя более активную роль в укреплении психического здоровья своих сотрудников. Лично меня воодушевляет число руководителей, с которыми я общалась и которые заинтересованы в наращивании экспертного опыта и потенциала в своих организациях в целях решения проблем психического здоровья сотрудников на рабочем месте.
Продолжая разбираться с неясными моментами и последствиями пандемии COVID-19, мы должны сохранять приверженность делу повышения осведомленности о психическом здоровье и настойчиво продвигать инновации для решения проблем, с которыми мы сталкиваемся как отдельные люди и как члены глобального общества. Мы должны продолжать расширять доступ к важнейшим услугам, которые могут дать людям возможность справиться с текущим кризисом и разрешить любые проблемы, которые могут возникнуть в будущем. Способствовать продолжению этого диалога и дальнейшему прогрессу – дело каждого из нас.
Примечание:
1Ronald C Kessler and others, “Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication”, Archive of General Psychiatry, vol. 62, No. 6 (June 2005), p. 600.
«Хроника ООН» не является официальным документом. Для нас большая честь публиковать статьи высокопоставленных лиц Организации Объединённых Наций, а также видных государственных и общественных деятелей со всего мира. Выраженные в статьях взгляды и мнения принадлежат авторам и могут не совпадать с официальной позицией Организации Объединённых Наций. Подобным образом указанные в статьях, картах и приложениях границы, географические названия и обозначения могут отличаться от официально признанных Организацией.
(PDF) ОПРЕДЕЛЯЮЩАЯ РОЛЬ КАТАЛИЗАТОРОВ В ПРОЦЕССЕ НИЗКОТЕМПЕРАТУРНОГО РАЗЛОЖЕНИЯ СЕРОВОДОРОДА: НЕРАВНОВЕСНАЯ ТЕРМОДИНАМИКА ОТКРЫТОЙ СИСТЕМЫ
Отличительными чертами термодинамики неравновесных процессов
является то, что рассматриваемые ею системы открыты для потоков вещества и
энергии, а процессы, протекающие в открытых системах, далеки от равновесия и
имеют необратимый характер. В термодинамике необратимых процессов важным
В термодинамике необратимых процессов важным
понятием является стационарное состояние системы. В отличие от
термодинамического равновесия, стационарное состояние характеризуется
постоянным притоком веществ в систему и удалением продуктов реакции,
непрерывной затратой свободной энергией, которая поддерживает постоянство
концентраций веществ в системе, и постоянством термодинамических параметров
(включая внутреннюю энергию и энтропию). Важно, что открытая система может
существовать только за счет притока энергии извне и оттока энергии в
окружающую среду [1-3].
Основной задачей термодинамики необратимых процессов является
определение величины прироста и оттока энтропии в реакционной системе на
основе уравнения Гиббса [1]. При стационарном неравновесном процессе в
открытой системе положительное производство энтропии внутри системы
компенсируется оттоком энтропии в окружающую среду, другими словами,
неравновесное состояние «сбрасывает» энтропию, выработанную внутри
системы, в окружающую среду, поддерживая этим стационарное состояние.
Стационарное неравновесное состояние образуется только в закрытых или
открытых системах. В изолированной неравновесной системе стационарное
состояние создать невозможно, так как с внешней средой система ничем не
связана, а внутренние самопроизвольные процессы обеспечивают только рост
энтропии вплоть до максимального Smax значения, переводя систему в
стационарное равновесное состояние.
Изменение энтропии открытой системы происходит как за счет ее
увеличения вследствие самопроизвольного протекания необратимых процессов
внутри самой системы (diS), так и за счет процессов обмена системы с внешней
средой (deS) [2]. В термодинамике необратимых процессов постулируют, что
составляющие diS и deS являются независимыми, а общее изменение энтропии
открытой системы равно их сумме: dS = diS + deS.
В открытых биологических системах наблюдается самопроизвольное
уменьшение энтропии даже при наличии в них необратимых процессов [7].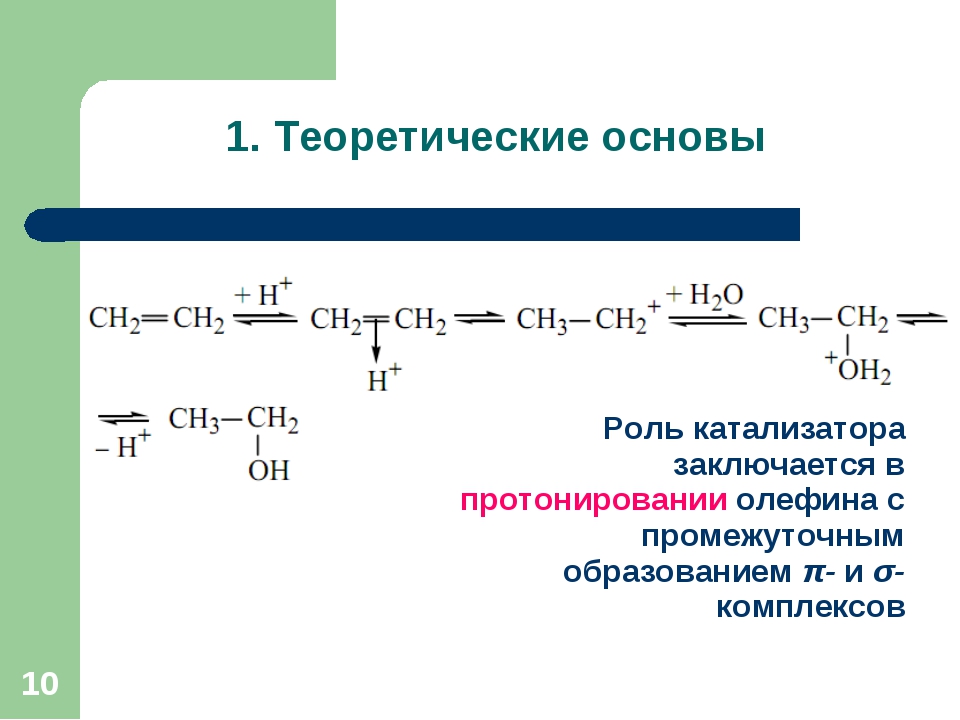 Это
Это
явление описывается [2] в рамках сопряжения химических процессов, приводящее
к увеличению энтропии окружающей среды. В частности, сопряжение двух
каталитических реакций может происходить за счет образования на поверхности
катализатора общих для них интермедиатов. В результате становится возможным
реализация брутто-реакции, формально приводящей к уменьшению энтропии [2].
В то же время, в биологических системах основным источником энергии для
обеспечения жизнедеятельности организмов является потенциальная энергия,
запасенная в химических связях [7,24]. Во всех клетках, энергия, выделившаяся в
результате разрыва определенных химических связей, используется для
генерирования потенциальной энергии в форме новых химических связей и
градиента концентрации. В соответствии с уравнением Гиббса G = H – TS,
самопроизвольное протекание необратимой химической реакции обеспечивается
при условии, что в экзотермическом процессе (энтальпия реакции уменьшается,
ΔН < 0) энтропия возрастает (ΔS > 0). Однако если в результате экзотермической
Однако если в результате экзотермической
реакции (ΔН < 0) энтропия уменьшается (ΔS < 0), то уравнение Гиббса определяет
границы самопроизвольного протекания процесса. При этом если некоторое
Каталитический риформинг бензинов, установка и гидроочистка бензиновых фракций
(cправочная информация)
Процесс каталитического риформинга бензиновых фракций (риформинга бензинов) является одним из важнейших процессов современной нефтеперерабатывающей и нефтехимической промышленности. Процесс риформинга предназначен для производства высокооктановых компонентов автомобильных бензинов и для производства легких ароматических углеводородов – бензола, толуола и ксилолов. Весьма важным продуктом процесса риформинга является водородсодержащий газ с высоким содержанием водорода, который используется для гидроочистки широкого ассортимента нефтяных фракций, для процесса гидрокрекинга тяжелых нефтяных фракций и других гидрогенизационных процессов.
Процесс каталитического риформинга является сложным химическим процессом. Это обусловлено, прежде всего, химическим составом исходного сырья процесса – разнообразных бензиновых фракций. В состав так называемой широкой фракции бензина входит более 150 углеводородов. Это углеводороды трех основных групп: парафиновые углеводороды нормального и изостроения, нафтеновые углеводороды с пятичленными и шестичленными циклами с одной или несколькими замещающими алкильными группами и ароматические углеводороды, которые обычно представлены бензолом, толуолом, ксилолами и незначительным количеством более тяжелых алкилбензолов. Среди парафинов преобладают углеводороды нормального строения и монометилзамещенные структуры. Нафтены представлены гомологами циклопентана и циклогексана.
Научные основы процесса каталитического риформинга были подготовлены работами русских учёных. Так ещё в 1911г. Н.Д. Зелинским была показана возможность дегидрогенизации шестичленных нафтеновых углеводородов при температуре выше 300°С над платиновым и палладиевым катализаторами количественно, практически без побочных реакций. В том же году дегидрогенизацию нафтеновых углеводородов при контакте их с оксидом металлов осуществили В.Н. Ипатьев и Н. Довгелевич. В 1936г. в СССР одновременно в трёх лабораториях была открыта реакция непосредственной дегидроциклизации парафиновых углеводородов в ароматические. Б.Л. Молдавский и Г.Д. Камушер в ГИВДс осуществили дегидроциклизацию парафинов на оксиде хрома при температуре 450-470°С. В.И. Каржёв, М.Г. Северьянова и А.Н. Сиова во ВНИГИ наблюдали реакции дегидроциклизации парафинов на меднохромовом катализаторе при температуре 500-550°С. Б.А. Казанский и А.Ф. Платэ в МГУ показали возможность дегидроциклизаци парафиновых углеводородов в присутствии платинированного угля при температуре 300-310°С.
В том же году дегидрогенизацию нафтеновых углеводородов при контакте их с оксидом металлов осуществили В.Н. Ипатьев и Н. Довгелевич. В 1936г. в СССР одновременно в трёх лабораториях была открыта реакция непосредственной дегидроциклизации парафиновых углеводородов в ароматические. Б.Л. Молдавский и Г.Д. Камушер в ГИВДс осуществили дегидроциклизацию парафинов на оксиде хрома при температуре 450-470°С. В.И. Каржёв, М.Г. Северьянова и А.Н. Сиова во ВНИГИ наблюдали реакции дегидроциклизации парафинов на меднохромовом катализаторе при температуре 500-550°С. Б.А. Казанский и А.Ф. Платэ в МГУ показали возможность дегидроциклизаци парафиновых углеводородов в присутствии платинированного угля при температуре 300-310°С.
Основой процесса каталитического риформинга бензинов являются реакции, приводящие к образованию ароматических углеводородов. Это реакции дегидрирования шестичленных и дегидроизомеризации пятичленных нафтеновых углеводородов, дегидроциклизация парафиновых углеводородов. Кроме того, второй по значимости в процессе каталитического риформинга является реакция изомеризации углеводородов.
Кроме того, второй по значимости в процессе каталитического риформинга является реакция изомеризации углеводородов.
Наряду с изомеризацией пятичленных и шестичленных нафтенов изомеризации подвергаются парафиновые и ароматические углеводороды. Существенную роль в процессе играют реакции гидрокрекинга парафинов, сопровождающиеся газообразованием. При каталитическом риформинге протекают также реакции раскрытия пятичленного кольца нафтенов с образованием соответствующих парафиновых углеводородов.
Типы установок риформинга бензиновых фракций
В настоящее время трудно найти завод, технология переработки нефти на котором не предусматривала бы каталитического риформирования. Развитие процесса каталитического риформинга было обусловлено длительной тенденцией роста октановых чисел товарных бензинов на фоне постепенного отказа от использования тетраэтилсвинца, как октаноповышающей добавки, а также ростом спроса на ароматические углеводороды. Таким образом, каталитический риформинг прочно занял место базового процесса современной нефтепереработки.
Эволюция процесса состояла в увеличении глубины превращения сырья, селективности ароматизации углеводородов и стабильности работы катализаторов. За весь период использования процесса выход ароматических углеводородов и водорода (целевые продукты) увеличился более чем в 1,5 раза, а межрегенерационный цикл работы катализатора — в 4 раза. Эти результаты достигнуты, прежде всего, за счет разработки новых катализаторов, повлекших за собой совершенствование технологии процесса. Сменилось, по меньшей мере, три поколения катализаторов, непременным компонентом которых всегда оставалась платина. Прогресс в технологии процесса выразился в снижении рабочего давления более чем в 10 раз (с 4,0 до 0,35 МПа) и разработке нового типа реакторных устройств непрерывного риформинга (системы CCR).
Технологическое оформление процесса каталитического риформинга определяется по способу проведения регенерации катализатора. Подавляющее большинство установок риформинга описывают тремя разновидностями технологий: полурегенеративный, циклический и процесс с непрерывной регенерацией катализатора. Наибольшее количество установок работает по полурегенеративному варианту. Например, платформинг фирмы ЮОП лицензирован примерно на 600 установках, магнаформинг фирмы Энгельгард осуществляется более чем на 150 установках, процесс ренийформинг фирмы Шеврон используется более чем на 70 установках, наконец, технология Французского института нефти лицензирована более чем на 60 установках мира. В России практически все установки каталитического риформинга (за исключением трех – в Уфе, Нижнем Новгороде и Омске) работают в полурегенеративном варианте.
Наибольшее количество установок работает по полурегенеративному варианту. Например, платформинг фирмы ЮОП лицензирован примерно на 600 установках, магнаформинг фирмы Энгельгард осуществляется более чем на 150 установках, процесс ренийформинг фирмы Шеврон используется более чем на 70 установках, наконец, технология Французского института нефти лицензирована более чем на 60 установках мира. В России практически все установки каталитического риформинга (за исключением трех – в Уфе, Нижнем Новгороде и Омске) работают в полурегенеративном варианте.
Технологические параметры работы установок риформинга по полурегенеративному варианту: давление- от 1.3 до 3.0 МПа, температура- от 480 до 530?С, октановое число (ИОЧ) колеблется от 94 до 100, выход риформата от 80 до 88% мас. Межрегенерационный цикл работы катализатора составляет от года до трех лет.
Второй тип технологии – циклический – применяется в основном на заводах США и характеризуется более жесткими условиями проведения процесса (давление 0. 9-2.1 МПа, температура 505-550?С) и, как следствие, небольшими межрегенерационными циклами (от 40 до 5 суток). Октановое число риформата (ИОЧ) – от 95 до 103. Катализатор до полной отработки может выдерживать до 600 регенераций. К циклическому варианту относится процесс пауэрформинг фирмы Эксон (около 100 установок) и ультраформинг фирмы Амоко Ойл Ко (~150 установок).
9-2.1 МПа, температура 505-550?С) и, как следствие, небольшими межрегенерационными циклами (от 40 до 5 суток). Октановое число риформата (ИОЧ) – от 95 до 103. Катализатор до полной отработки может выдерживать до 600 регенераций. К циклическому варианту относится процесс пауэрформинг фирмы Эксон (около 100 установок) и ультраформинг фирмы Амоко Ойл Ко (~150 установок).
Наконец, третий тип технологии каталитического риформинга представляет собой процесс с непрерывной регенерацией катализатора. Данная технология наиболее прогрессивна, так как позволяет работать в лучших термодинамических условиях (давление – 0.35-0.9 МПа, температура –до 550?С) без остановки на регенерацию (межремонтный пробег установок риформинга достигает 3-х лет и более) и достигнуть максимального октанового числа риформата (ИОЧ=102-104).
Первая установка запущена по лицензии фирмы ЮОП в 1971 году, в 1983году эксплуатировалось 35 установок, а в настоящее время работает 163 установки (в том числе 40 с давлением 0,35 МПа) по лицензии ЮОП и 56 установок по лицензии Французского института нефти.
Классификация промышленных установок риформинга
В России подавляющее большинство установок каталитического риформинга относится к классу полурегенеративного типа. Установки каталитического риформинга состоят из двух блоков. На первой стадии исходное сырье подвергается предварительной гидроочистке бензиновых фракций с целью практически полного удаления присутствующих в нем примесей органических соединений серы, азота, кислорода, хлора и др., являющихся ядами для катализаторов, используемых в процессе каталитического риформинга. На второй стадии гидроочищенное сырье подвергается непосредственно каталитическому риформингу.
Упрощенная принципиальная схема блока каталитического риформинга представлена на рис. 1.
Сырье – стабильный гидрогенизат с блока, где происходит гидроочистка бензиновых фракций поступает на прием сырьевого насоса Н-1, который подает его в тройник смешения на смешение с циркулирующим водородсодержащим газом (ВСГ), поступающим с выкида циркуляционного компрессора ЦК-1. Смесь сырья и ВСГ в теплообменнике Т-1 подогревается газопродуктовым потоком, выходящим из реактора Р-3, подогревается в первой секции печи П-1 и поступает в реактор Р-1, затем подогревается во второй секции П-1, проходит реактор Р-2, затем проходит третью секцию печи П-1 и проходит в реактор Р-3. Газопродуктовая смесь после реактора Р-3 отдает часть своего тепла газосырьевому потоку в теплообменнике Т-1, охлаждается в воздушном холодильнике ВХ-1, в водяном холодильнике Х-1 и поступает в газосепаратор С-1. Здесь происходит отделение водородсодержащего газа от жидкого продукта – нестабильного катализата. Водородсодержащий газ из сепаратора С-1 направляется на удаление избыточной влаги в адсорбер А-1 (или минует его по байпасу) и поступает на прием циркуляционного компрессора ЦК-1, который вновь подает его на смешение с сырьем.
Смесь сырья и ВСГ в теплообменнике Т-1 подогревается газопродуктовым потоком, выходящим из реактора Р-3, подогревается в первой секции печи П-1 и поступает в реактор Р-1, затем подогревается во второй секции П-1, проходит реактор Р-2, затем проходит третью секцию печи П-1 и проходит в реактор Р-3. Газопродуктовая смесь после реактора Р-3 отдает часть своего тепла газосырьевому потоку в теплообменнике Т-1, охлаждается в воздушном холодильнике ВХ-1, в водяном холодильнике Х-1 и поступает в газосепаратор С-1. Здесь происходит отделение водородсодержащего газа от жидкого продукта – нестабильного катализата. Водородсодержащий газ из сепаратора С-1 направляется на удаление избыточной влаги в адсорбер А-1 (или минует его по байпасу) и поступает на прием циркуляционного компрессора ЦК-1, который вновь подает его на смешение с сырьем.
Избыток ВСГ направляется на блок гидроочистки бензиновых фракций или в водородное кольцо завода. Нестабильный катализат из сепаратора С-1 подогревается в теплообменнике Т-2 потоком стабильного катализата и поступает в среднюю часть колонны К-1 на стабилизацию – отделение растворенных в нем газообразных углеводородов. Верхом колонны К-1 выводятся легкие углеводороды до бутанов включительно. Пары охлаждаются и конденсируются в воздушном холодильнике ВХ-2 и водяном холодильнике Х-2 и поступает в емкость орошения Е-1. Жидкий продукт из Е-1 поступает на прием насоса Н-2, который подает его в качестве холодного орошения на верхнюю тарелку колонны К-1. Балансовый избыток выводится на ГФУ или в парк в виде жидкого газа. Несконденсировавшиеся газы из емкости Е-1 сбрасываются в топливную сеть.
Верхом колонны К-1 выводятся легкие углеводороды до бутанов включительно. Пары охлаждаются и конденсируются в воздушном холодильнике ВХ-2 и водяном холодильнике Х-2 и поступает в емкость орошения Е-1. Жидкий продукт из Е-1 поступает на прием насоса Н-2, который подает его в качестве холодного орошения на верхнюю тарелку колонны К-1. Балансовый избыток выводится на ГФУ или в парк в виде жидкого газа. Несконденсировавшиеся газы из емкости Е-1 сбрасываются в топливную сеть.
Стабильный катализат риформинга выводится снизу колонны К-1, проходит теплообменник Т-2, охлаждается в воздушном холодильнике ВХ-3, водяном холодильнике Х-3 и направляется в парк в качестве готового продукта. Подвод тепла в низ колонны К-1 осуществляется циркуляцией части стабильного катализата через печь П-2 под нижнюю тарелку колонны. Для компенсации уноса части хлора с поверхности катализатора схемой предусмотрена дозированная подача раствора хлорорганического соединения на вход первого либо в каждый из реакторов. Для поддержания водно-хлорного баланса в зоне катализа предусматривается дозированная подача воды в реакторный блок, включая возможность подачи отдельно в каждый реактор.
Для поддержания водно-хлорного баланса в зоне катализа предусматривается дозированная подача воды в реакторный блок, включая возможность подачи отдельно в каждый реактор.
Для выполнения операции осернения катализатора в пусковой период схемой предусматривается дозированная подача в реакторный блок раствора сероорганического соединения. Схемой предусмотрена также подача в каждый реактор хлорорганического соединения для выполнения операции реактивации катализатора риформинга.
Таблица 2. Установки риформинга для производства бензина (по типовым проектам)
Установки каталитического риформинга, предназначенные для производства компонента автомобильного бензина, состоят из двух основных блоков – блока, где происходит гидроочистка бензиновых фракций и блока риформинга. Исключением является установка Л-35-5/300, которая, являясь первенцем промышленных установок риформинга, первоначально была спроектирована в виде самостоятельного блока каталитического риформинга. Эта установка работает в комплексе с отдельно стоящей типовой установкой гидроочистки Л-24-300.
Эта установка работает в комплексе с отдельно стоящей типовой установкой гидроочистки Л-24-300.
Таблица 3. Установки каталитического риформинга для производства ароматических углеводородов (по типовым проектам)
Значительно более сложный технологический комплекс представляет собой установка каталитического риформинга, предназначенная для производства ароматических углеводородов. В этот комплекс кроме блока гидроочистки и блока риформинга входит также блок экстракции ароматических углеводородов из катализата риформинга и блок четкой ректификации для разделения ароматического экстракта с получением ароматических углеводородов товарного качества. В табл. 2. представлена краткая характеристика основных типов установок риформинга, предназначенных для производства компонента автомобильного бензина. В табл. 3. представлена краткая характеристика типовых установок риформинга, предназначенных для производства ароматических углеводородов. Представленные данные характеризуют установки по материалам типовых проектов.
Таблица 4. Объем системы установок риформинга
В табл. 4 даны объемы систем блоков гидроочистки и риформинга. Эти данные необходимы для расчетов расхода водородсодержащего газа и технического азота на период пуска установок и регенерации катализатора. Принципиальные технологические схемы блока, где происходит гидроочистка бензиновых фракций и риформинга практически идентичны. Но имеются некоторые отличия, которые заключаются в основном в аппаратурном оформлении отдельных узлов установок, прежде всего, узлов стабилизации нестабильного катализата риформинга. Для установок, предназначенных для производства высокооктанового компонента автобензина характерно увеличение производственной мощности по перерабатываемому сырью с 300 тыс. т/год до 600 тыс. т/год и до 1000 тыс. т/год, что диктовалось необходимостью увеличения производства высокооктановых автомобильных бензинов. Все установки, предназначенные для производства ароматических углеводородов, имели одинаковую производительность – 300 тыс. т/год по сырью. Установки, рассчитанные на переработку высоконафтенистого сырья, имели реакторные узлы, состоящие из четырех реакторов – четырех ступеней реакции. Это установки типа Л-35-12/300, Л-35-12/300А и Л-35-13/300А. Остальные установки этого рода имели реакторные узлы из трех ступеней реакции.
т/год по сырью. Установки, рассчитанные на переработку высоконафтенистого сырья, имели реакторные узлы, состоящие из четырех реакторов – четырех ступеней реакции. Это установки типа Л-35-12/300, Л-35-12/300А и Л-35-13/300А. Остальные установки этого рода имели реакторные узлы из трех ступеней реакции.
Катализаторы риформинга
В процессе каталитического риформинга используются катализаторы, основой которых является платина, равномерно распределенная на носителе – оксиде алюминия, промотированном хлором (в редких случаях фтором). Природа активной поверхности катализаторов риформинга базируется на модели бифункционального их действия, предложенной в 1953г. Маилсом. Диспергированная на поверхности носителя платина является катализатором реакций гидрирования-дегидрирования, а носитель – галоидированный оксид алюминия – катализатором реакций кислотно-основного типа – изомеризации, циклизации, крекинга.
Новейшими исследованиями, выполненными в последнее время, было обнаружено, что часть высокодисперсной нанесенной на носитель платины по своим физическим, адсорбционным и химическим характеристикам не соответствует характеристикам металлической платины. Эта платина получила название электронодефицитной и обозначается символом Ptσ в отличие от металлической платины, которая обозначается символом Pt?. Характерной особенностью электронодефицитной платины является ее способность образовывать прочную хемосорбционную связь с молекулами воды. По этому признаку все поверхностные атомы платины на катализаторе различаются на два состояния: Pt? и Ptσ. Эта же характерная особенность электронодефицитной платины позволяет оценивать ее количество на поверхности катализатора.
Эта платина получила название электронодефицитной и обозначается символом Ptσ в отличие от металлической платины, которая обозначается символом Pt?. Характерной особенностью электронодефицитной платины является ее способность образовывать прочную хемосорбционную связь с молекулами воды. По этому признаку все поверхностные атомы платины на катализаторе различаются на два состояния: Pt? и Ptσ. Эта же характерная особенность электронодефицитной платины позволяет оценивать ее количество на поверхности катализатора.
Главной характерной особенностью электронодефицитной платины Ptσ является ее высокая активность в реакции дегидроциклизации парафиновых углеводородов – основополагающей реакции процесса каталитического риформинга бензиновых фракций. Скорость реакции дегидроциклизации парафиновых углеводородов с участием платины Ptσ в десять-пятнадцать раз выше скорости с участием металлической платины Pt?. Электронодефицитная платина Ptσ входит в состав поверхностных комплексов PtClxOyLz, являющихся продуктами сильного взаимодействия предшественника платины с поверхностными группами и дефектами γ- или η-оксидов алюминия,являющегося основным носителем катализаторов риформинга. Характерными признаками состояния Ptσ являются предельная дисперсность, ионные состояния платины, наличие лигандов L, связанных с носителем, отсутствие связи Pt-Pt,высокая устойчивость к спеканию. Установлена линейная зависимость между константой скорости дегидроциклизации парафинового углеводорода и содержанием платины Ptσ в катализаторе, что дает основание отнести Ptσ к активным центрам ароматизации парафинов, обладающих комплексом свойств, обуславливающих высокую активность и селективность действия в сложной реакции дегидроциклизации парафиновых углеводородов.
Характерными признаками состояния Ptσ являются предельная дисперсность, ионные состояния платины, наличие лигандов L, связанных с носителем, отсутствие связи Pt-Pt,высокая устойчивость к спеканию. Установлена линейная зависимость между константой скорости дегидроциклизации парафинового углеводорода и содержанием платины Ptσ в катализаторе, что дает основание отнести Ptσ к активным центрам ароматизации парафинов, обладающих комплексом свойств, обуславливающих высокую активность и селективность действия в сложной реакции дегидроциклизации парафиновых углеводородов.
Разработанные технологии приготовления современных катализаторов риформинга направлены на получение катализаторов с максимальным содержанием электронодефицитной платины Ptσ. Наиболее активные и стабильные современные промышленные катализаторы содержат в своем составе до 55 % Ptσ от общего содержания платины в катализаторе.
Большинство промышленных катализаторов риформинга приготовлено с использованием в качестве носителя γ-Al2O3, обладающей большей термической стабильностью.
Для усиления и регулирования кислотной функции оксид алюминия промотируют галоидом – фтором или хлором. Фторсодержащие катализаторы используются весьма ограниченно, в случаях, когда процесс риформинга осуществляют без предварительной гидроочистки сырья или при высокой влажности. Абсолютное большинство катализаторов риформинга приготовлены на основе хлорированного оксида алюминия. Преимуществом катализаторов, приготовленных на хлорированном оксиде алюминия, является возможность регулирования содержания хлора на поверхности катализаторов, а, следовательно, и уровень их кислотности, непосредственно в условиях эксплуатации. Это объясняется тем, что хлор является подвижным промотором, он слабо связан с поверхностью носителя и легко замещается гидроксилами воды.
Количество хлора на поверхности оксида алюминия определяется равновесием реакции:
Это обстоятельство привело к необходимости во время эксплуатации поддерживать над поверхностью катализатора вполне определенную концентрацию паров воды, при которой в катализаторе содержится оптимальное количество хлора, и которое, как правило, находится в пределах 0,9-1,2 масс. %. Содержание хлора на поверхности катализатора является функцией мольного отношения вода: хлор в зоне реакции, удельной поверхности Al2О3 и прочности удерживания хлора на катализаторе.
%. Содержание хлора на поверхности катализатора является функцией мольного отношения вода: хлор в зоне реакции, удельной поверхности Al2О3 и прочности удерживания хлора на катализаторе.
Высока роль хлора в создании активной поверхности катализатора, в создании поверхностных комплексов, обеспечивающих стабильную работу катализаторов в жестких условиях процесса. Поверхностные комплексы имеют примерный состав PtσnClxOyLz, где σ=2; n≥1; x+y+z≤4; в качестве лигандов L могут быть ионы S, углеводородные радикалы (влияние реакционной среды).
Наконец, без хлора невозможно восстановление высокой дисперсности платины на носителе в период реактивации платиновых катализаторов.
В настоящее время в промышленной практике используются модифицированные би- и полиметаллические катализаторы риформинга, приготовленные на хлорированном оксиде алюминия, в которых наряду с платиной содержатся другие элементы периодической системы. Модификаторами для катализаторов риформинга являются рений, олово, титан, германий, иридий, свинец, цирконий, марганец.
Основным преимуществом модифицированных полиметаллических катализаторов риформинга является их высокая стабильность, выражающаяся в том, что снижение активности в условиях процесса происходит значительно медленнее, чем у монометаллических платиновых катализаторов.
Поскольку основной причиной дезактивации катализаторов риформинга в цикле реакции является их закоксовывание, повышение стабильности при введении модифицирующих металлов связано с воздействием на процесс коксоотложения. Характер этого воздействия, его механизм зависит от природы применяемого модификатора.
В промышленной практике процесса риформинга наибольшее распространение получили алюмоплатиновые катализаторы, модифицированные рением – платинорениевые катализаторы, в отдельных случаях с добавками третьего компонента.
Информация данного раздела приведена исключительно в справочных целях. Информацию о продукции и услугах ООО «НПП Нефтехим» Вы найдете в разделах Главное меню/Разработки и Услуги.
Роснефть начала производство инновационного катализатора нового поколения
Москва, 16 янв — ИА Neftegaz.RU. Специалисты РН-ЦИР разработали катализатор гидроочистки дизельных фракций Ht-100RN, который обеспечивает выработку дизтоплива стандарта Евро-5, и по своим эксплуатационным свойствам превосходит зарубежные аналоги.Об этом сообщила Роснефть.
Компании удалось реализовать полный цикл создания инновационного продукта: от научной разработки до промышленных испытаний.
Производство катализатора запущено на Ангарском заводе (АЗКиОС), а опытно-промышленная эксплуатация проводится на Башнефть-Уфанефтехим.
АЗКиОС является специализированным предприятием по производству широкого спектра катализаторов, адсорбентов, носителей для катализаторов, осушителей и цеолитов, а также продукции органического синтеза в виде присадок и добавок к моторному топливу.
Продукция завода зарекомендовала себя на нефтеперерабатывающих, химических, азотных, газоперерабатывающих, и др. заводах России ближнего и дальнего зарубежья.
заводах России ближнего и дальнего зарубежья.
По результатам испытаний подтверждены уникальные эксплуатационные свойства: Ht-100RN работает при более низкой температуре (на 5-10 C ниже, чем аналоги), характеризуется высокой стабильностью эксплуатационных показателей и более продолжительным сроком работы — в 2-3 раза превышающем срок эксплуатации импортных аналогов.
Промышленные испытания Ht-100RN начались в августе 2018 г., на текущий момент промышленный пробег катализатора продолжается 2й год без перегрузки и регенерации.
О завершении испытаний глава Роснефти И. Сечин доложил премьер-министру РФ Д. Медведеву на встрече в правительственной резиденции Горки 1 октября 2019 г.
Подробно на этой теме в ходе встречи Д. Медведев и И. Сечин не останавливались, отметив важность этого производства, поскольку все катализаторы ранее приобретались за границей.
И. Сечин отметил, что новое производство способно произвести объем катализаторов, который необходим для России в целом.
Т.е. речь идет не только об обеспечении потребностей предприятий Роснефти в катализаторах процессов гидроочистки, но и поставках для коммерческой реализации.
По итогам проведенных испытаний принято решение о запуске серийного производства нового инновационного катализатора на заводах компании с его последующей загрузкой на НПЗ Роснефти.
Ht-100RN стал 1м отечественным катализатором гидроочистки дизтоплив, внедренным в промышленное производство в России за последние 20 лет.
Роснефть в рамках импортозамещения уделяет особое внимание развитию собственного производства катализаторов для нефтепереработки.
В 2018 г. Роснефть начала использовать катализаторы Ангарского завода катализаторов и органического синтеза на установках по производству водорода на Куйбышевском НПЗ и Рязанской НПК.
Ранее такие же ангарские катализаторы заменили импортные аналоги парового риформинга метана на водородных установках Башнефть-Уфанефтехим, Башнефть-Новойл и Сызранского НПЗ.
Доля собственных катализаторов парового риформинга при производстве водорода на НПЗ Роснефти достигла 77%.
Роль катализаторов в нефтепереработке велика, что вынуждает ведущие российские компании заниматься разработкой импортозамещающей продукции.
В России активно занимается развитием катализаторного бизнеса Газпром нефть, для которой это стратегическое направление и приоритетный проект сегмента downstream.
Проект Газпром нефти по развитию производства катализаторов имеет статус нацпроекта и является прорывным не только для нефтепереработки.
Подробнее читайте в спецпроекте Neftegaz.RU «Национальный продукт: отечественные катализаторы», разработанный совместно с Газпром нефтью.
Катализаторы реакции гидролиза
Реакции целлюлозы могут быть гомогенными и гетерогенными, т. е. протекать в гомогенной и гетерогенной средах. В гомогенных реакциях реагирует растворенная целлюлоза. Примером гомогенной реакции может служить гидролиз целлюлозы в 72%-ной серной кислоте. Целлюлоза сначала растворяется в такой кислоте, а затем уже гидролизуется. Реакция ацетилирования целлюлозы уксусным ангидридом в ледяной уксусной кислоте или в метиленхлориде в присутствии катализатора начинается в гетерогенной среде, а заканчивается в гомогенной среде. В результате гомогенных реакций получаются продукты, более однородные по свойствам и по химическому составу (по степени замещения), чем при реакциях гетерогенных. Большинство процессов этерификации и других реакций целлюлозы относится к гетерогенным реакциям.[ …]
Гидролиз полисахаридов растительной ткани в холодной воде практически не наблюдается. При повышении температуры воды выше 100° гидролиз полисахаридов протекает, но настолько медленно, что практического значения такой процесс не имеет. Удовлетворительные результаты получаются только при применении катализаторов, из которых производственное значение имеют лишь сильные минеральные кислоты: серная и реже соляная. Чем выше концентрация сильной кислоты в растворе и температура реакции, тем быстрее протекает гидролиз полисахаридов до моносахаридов. Однако присутствие таких катализаторов имеет и отрицательную сторону, так как они одновременно с реакцией гидролиза полисахаридов ускоряют и реакции распада моносахаридов, соответственно снижая этим их выход.[ …]
При гидролизе гликозидных связей в качестве катализаторов реакции действуют хлор и ион водорода. Был предложен и механизм протекания этих реакций [47]. Отмечено было также, что гидроксильные группы пиранозного кольца, находящиеся в положении 2 и 6, могут в указанных выше условиях окисляться до карбоксильных групп. Все эти реакции могут протекать при хлорировании и отбелке целлюлозы, однако масштабы этих реакций недостаточно ясны. В литературе имеются указания на то, что эти реакции протекают значительно медленнее хлорирования лигнина и отбелки [47], вследствие чего их роль незначительна.[ …]
Вода — катализатор многих химических реакций, и иногда для прохождения реакции необходимы хотя бы ее следы. Взаимодействуя с некоторыми солями, вода вызывает процесс обменного разложения их — гидролиз. Вода — участник и среда для протекания множества биохимических реакций в живых организмах.[ …]
Во многих реакциях вода служит катализатором, а иногда, напротив, — каталитическим ядом. Растворяя окислы, она образует кислоты или щелочи. При взаимодействии ее с солями слабых кислот или слабых оснований происходит гидролиз этих солей. Растворяя какие-либо вещества, вода в ряде случаев образует гидраты — более или менее прочные соединения частиц растворенного вещества с молекулами воды.[ …]
Поскольку гидролиз макромолекул гемицеллюлоз протекает постепенно, с разрывом равноценных связей в скелетах ксиланов, глюкоманнанов или галактанов, в первых стадиях реакции образуются крупные обломки макромолекул с постепенно уменьшающейся степенью полимеризации. По мере развития реакции гидролиза эти обломки постепенно уменьшаются и доходят в конечном итоге до соответствующих моносахаридов. Поскольку реакция гидролиза полисахаридов протекает в водной среде, в присутствии иона водорода, который должен иметь доступ к разрываемым гли-козидным связям, в первую очередь гидролизу подвергаются перешедшие в раствор макромолекулы гемицеллюлоз. При этом необходимо отметить, что в растворе могут оказаться отдельные макромолекулы, легко подвергающиеся гидролизу, поскольку катализатор свободно подходит к ним со всех сторон, и сгустки макромолекул, образующие коллоидные частицы или мицеллы различных размеров. Последние вследствие затрудненного доступа катализатора в их толщу гидролизуются несколько медленнее [9], причем константы скорости их гидролиза могут различаться между собой в 2—3 раза.[ …]
Совершенно иначе эта реакция протекает в присутствии катализаторов, направляющих ее в сторону образования моносахаридов. Катализаторами являются кислоты, кислые соли и другие соединения, которые в водном растворе диссоциируют с образованием ионов водорода. Последний собственно и является катализатором гидролиза гликозидных связей. Каталитическая активность веществ, образующих в водном растворе ионы водорода, зависит от их силы или способности диссоциировать. Чем полнее вещество диссоциирует в воде, тем более сильным катализатором оно является. Так, кислоты по их каталитической активности образуют ряд, соответствующий их силе диссоциации. В этом ряду на первом месте стоят минеральные кислоты, на 100% диссоциирующие в разбавленных водных растворах, например соляная кислота. Каталитическая активность серной кислоты почти в 2 раза меньше, так как ее второй водород имеет значительно меньшую константу диссоциации.[ …]
Винилалкиловые эфиры гидролизуются с образованием ацетальдегида и спирта, иногда при комнатной температуре в присутствии небольшого количества кислоты. Способны к реакциям присоединения спиртов, органических кислот, галогенов, к конденсации с ароматическими соединениями и диеновыми углеводородами, к полимеризации в присутствии кислых катализаторов (хлористого и бромистого водорода, хлорного железа и др.). При полимеризации виниловых эфиров образуются высокомолекулярные продукты.[ …]
Наиболее легко подвергаются гидролизу гетероцепные полимеры. По способности к гидролизу их можно расположить в следующий ряд: полисахариды>полиамиды (белки и синтетические полиамиды) >сложные полиэфиры>простые полиэфиры. Катализаторами реакции гидролиза служат ионы водорода Н+ и гидроксила ОН-. Ионы водорода (кислоты) сильнее разрушают полисахариды (например, хлопчатобумажные ткани), а ионы гидроксила (щелочи) — белки (например, шерстяные ткани). Катализаторами гидролиза природных гетеро-цепных полимеров могут быть также различные ферменты. Реакцию гидролитической деструкции применяют специально для получения простых сахаров (моносахаридов) из полисахаридов и в том числе из полисахаридов древесины в гидролизном производстве (см. с. 123).[ …]
Эффективность общего катализа реакции гидролиза эфира оценивается по возрастанию константы скорости реакции гидролиза при увеличении концентрации кислоты или основания. Обычно это делается при постоянном значении pH, что обеспечивает постоянство отношения кислых и основных форм катализатора. Важно, чтобы ионная сила реакционной смеси сохранялась постоянной, поскольку многие реакции чувствительны к изменению концентрации солей. Чтобы выяснить, какой формой катализатора — кислой или основной — осуществляется катализ, необходимо провести измерения, меняя состав буфера. Скорость гидролиза эфира обычно пропорциональна концентрации основного компонента буфера, т. е. имеет место общий основный катализ. Наклон прямой, представляющей собой зависимость константы скорости от концентрации основания, дает константу скорости второго порядка для общего основного катализа (рис. 2.3).[ …]
На основании изучения кинетики гидролиза диэпоксисоединений (di- и мезо-изомеров) в присутствии Н+-ионов найдены оптимальные условия сшивания целлюлозы диэпоксибутадиеном. Установлены также оптимальные соотношения диэпоксисоединения, воды и катализатора при проведении реакции сшивания в присутствии фторбората цинка [100, 101]. Получены сшитые продукты сложного состава при взаимодействии целлюлозы с 1,3-диглицидил-глицерином в присутствии большого числа катализаторов, в том числе кислотных [102]. Обработка целлюлозных материалов N,N,N,-t/)uc- (2,3-эпоксипропил) амином и N,N,N-rpuc- (З-хлор-2-ок-силпропил) амином в присутствии щелочи привела к получению продуктов сложного состава, которые не были достаточно охарактеризованы [103]. Такую обработку можно проводить и в присутствии кислотных катализаторов, однако при этом процесс сшивания осложняется побочной реакцией полимеризации.[ …]
На основании изучения кинетики гидролиза диэпоксисоединений (di- и мезо-изомеров) в присутствии Н+-ионов найдены оптимальные условия сшивания целлюлозы диэпоксибутадиеном. Установлены также оптимальные соотношения диэпоксисоединения, воды и катализатора при проведении реакции сшивания в присутствии фторбората цинка [100, 101]. Получены сшитые продукты сложного состава при взаимодействии целлюлозы с 1,3-диглицидил-глицерином в присутствии большого числа катализаторов, в том числе кислотных [102]. Обработка целлюлозных материалов N,N,N,-t/)uc- (2,3-эпоксипропил) амином и N,N,N-rpuc- (З-хлор-2-ок-силпропил) амином в присутствии щелочи привела к получению продуктов сложного состава, которые не были достаточно охарактеризованы [103]. Такую обработку можно проводить и в присутствии кислотных катализаторов, однако при этом процесс сшивания осложняется побочной реакцией полимеризации.[ …]
Химические свойства. Наиболее характерная реакция — гидролиз (омыление). По способности гидролизоваться С. Э. занимают промежуточное положение между простыми эфирами и ангидридами. Многие С. Э. водой гидролизуются очень медленно. Катализаторами гидролиза являются кислоты (Н+-ионы). Особенно ускоряют гидролиз С. Э. щелочи, связывающие образующуюся при гидролизе кислоту. С. Э. — ацилирующие средства, менее энергичные, чем ангидриды.[ …]
Изофермент С является чрезвычайно эффективным катализатором. Для реакции гидратации Исах равна 106с-1, а /См для СОг — 8,3 мМ, в то время как для реакции дегидратации са4 = 6-105 с-1, а /См для НСОз равна 32 мМ [215]. Каталитическая активность фермента определяется состоянием ионизации группы с р/Са = 7 в свободном ферменте [215]. Реакция гидратации зависит от доли этих групп, находящихся в основной форме, а реакция дегидратации — от доли этих же групп, находящихся в кислой форме. Число оборотов для указанных реакций Значительно выше констант скорости переноса протонов между водой и группой с р/Са = 7 (приблизительно 2-103 с-1; табл. 4.2). В нефизиологических реакциях эти ферменты катализируют также гидратацию альдегидов и гидролиз эфиров.[ …]
Водорастворимая ацетилцеллюлоаа может быть получена путем глубокого гидролиза триацетилцеллюлозы в гомогенной среде; этот процесс подробно изучен в работах [238—241]. Триацетат целлюлозы в свою очередь получали путем взаимодействия хлопкового линтера и ангидрида уксусной кислоты в присутствии катализатора Н2804. После завершения растворения триацетата целлюлозы и образования вязкого прозрачного раствора последний подвергался нагреванию-при постоянной температуре (+0.1 К) при непрерывном перемешивании. При гидролизе триацетата целлюлозы происходит изменение его лиофильности. Для поддержания постоянной растворимости и проведения гидролиза к раствору в определенные промежутки времени добавлялась вода. В конечной стадии гидролиза концентрация уксусной кислоты в смеси не должна превышать 40 %. Контроль за течением реакции гидролиза осуществлялся путем титрования отдельных проб раствора метилэтилкетоном и водой [64, 239].[ …]
Из большого числа возможных вариантов концентрации кислоты и температуры реакции в настоящее время практически применяются только два: гидролиз разбавленными кислотами и гидролиз концентрированными кислотами. При гидролизе разбавленными кислотами температура реакции обычно составляет 160—190° и концентрация катализатора в водном растворе колеблется от 0,3 до 0,7% (Н2504, НС1).[ …]
Гидролитическая деструкция целлюлозы рассмотрена в [1]. С химической точки зрения гидролиз целлюлозы аналогичен гидролизу дисахаридов [1а]. В присутствии кислотных катализаторов происходит быстрое образование промежуточного комплекса между глюкозидным кислородом и протоном, приводящее к медленному, определяющему скорость реакции расщеплению глюко-зидной связи у С(1) [2]. При избытке воды реакция, как правило, подчиняется уравнению первого порядка; тем не менее изучение начальных стадий реакции гидролиза показало, что во многих случаях они могут быть описаны уравнением нулевого порядка.[ …]
На разложение пестицидов в почве оказывают влияние ее физические и химические свойства. Глины, оксиды, гидроксиды и ионы металлов при участии грунтовой воды служат катализаторами в этом процессе. Отмечено, что интенсивно гидролиз пестицидов происходит в почвах с сильнокислой реакцией и с высоким содержанием гумуса. Важную роль в процессе их разложения играют высшие растения и микроорганизмы — бактерии, актиномицеты, грибы.[ …]
Питательные вещества в бактериальную клетку поступают через всю поверхность тела и только в растворенном состоянии. Нерастворенные и коллоидные (эмульгированные) вещества могут предварительно переводиться в водорастворимые состояния с помощью особых катализаторов химических реакций — ферментов. Они вызывают гидролиз веществ до более простых и растворимых в воде соединений. Каждый фермент действует лишь па строго определенное вещество, и поэтому микроорганизм вырабатывает в себе комплекс разнообразных ферментов, соответствующих его физиологическим особенностям и потребностям.[ …]
При обычной температуре нейтральные водные растворы мочевины сравнительно стабильны и только под действием кислоты и щелочи при нагревании мочевина разлагается с образованием аммиака и диоксида углерода. Процесс этот рекомендован для очистки от мочевины сточных вод; он достаточно подробно изучен, особенно в рамках лабораторных исследований. В работе [92] рассмотрен щелочной гидролиз мочевины при содержании ее в растворе 1-50 г/дм3. Оптимальными условиями процесса являются молярные соотношения NaOH и (NHj CO — 1,5 :1, температура 140—155 °С, продолжительность реакции 15—30 мин. Сточные воды, содержащие после гидролизных установок до 100— 130 мг/дм3 аммиака и мочевины, могут быть при необходимости доочи-щены биохимическим способом [93]. Предложена интенсификация процесса гидролиза мочевины путем применения катализатора — соединений ванадия (пат. Реакцию проводят при температуре от 70 до 200 °С (оптимальная температура — 90—160 °С), при дозировке катализатора 0,02—1,0 % в пересчете на ванадий (оптимальное количество — 0,05—0,7 %). Степень гидролиза регулируют временем течения процесса, продолжительность которого изменяется от 10 до 360 мин.[ …]
Финансовый кризис сыграл роль катализатора процесса удешевления нефти
Публикации — ТЭК
Финансовый кризис сыграл роль катализатора процесса удешевления нефти. Однако резкий обвал произошел бы все равно, поскольку за последние годы в мировой экономике накопилась слишком большая психологическая усталость от неоправданно высоких цен на сырьевые товары, заявил для БГНЕС руководитель отдела исследований газовой отрасли ИПЕМ Алексей Белогорьев.
В последние годы происходил стремительный рост мировых цен на нефть, пик которого был зарегистрирован в начале июля 2008 г. Тогда баррель „черного золота» стоил рекордные 147,27 доллара. Шесть месяцев спустя ситуация поменялось в корне: нефть подешевела уже на 70%, а её цена пробила психологический барьер в 50 долларов за баррель. О факторах, которые привели к этим ценам, влиянии глобального финансового кризиса на нефтяной сектор и о влиянии всего этого на российскую экономику, рассказал в интервью для БГНЕС Алексей Белогорьев, руководитель Отдела исследований газовой отрасли Института проблем естественных монополий. В середине ноября он выступил с докладом „Перспективы динамики мировых цен на нефть и природный газ в условиях мирового финансового кризиса» на VI Международном форуме «ГАЗ РОССИИ-2008″, который состоялся в Москве. Алексей Белогорьев является экспертом по проблемам газа и нефти в России, Центральной Азии и Каспийском регионе, российским инвестициям на Балканском полуострове, газопроводу „Южный поток». Его экспертные оценки и публикации печатаются в ведущих газетах /РБК Дейли, Независимая газета/ и научных журналах /Мировая энергетика/.
Какие факторы привели к стремительному росту мировых цен на нефть, который мы наблюдали в последние годы? Какую роль в этом сыграл «спекулятивный пузырь» на сырьевых рынках и повсеместный экономический рост, прежде всего в таких странах как Китай и Индии?
У роста 2000-х гг. были как объективные, так и субъективные причины. К объективным я бы отнес, прежде всего, почти повсеместное удорожание себестоимости добычи углеводородов, а также острую необходимость масштабных инвестиций в новые нефтедобывающие проекты под угрозой возникновения дефицита на нефтяных рынках. Кроме того, большое влияние оказывал быстрый рост спроса на энергоресурсы со стороны развивающихся стран, прежде всего, Китая. Все указанные факторы актуальны и сегодня, в связи с чем падение цен до уровня 1990-х гг. практически исключено даже при очень негативном сценарии развития мирового кризиса.
Влияние этих объективных факторов подняло цены на нефть до уровня 80-90 долл. за барр. Весь последующий рост был исключительным детищем спекулятивного капитала, рассматривающего товарные рынки как удачное место для краткосрочных инвестиций. Не удивительно, что в последние годы физическая поставка нефти по биржевым контрактам происходила лишь примерно в 1% случаев. То есть 99% от всего объемов торгов были обычной перепродажей.
В последние месяцы мы стали свидетелями противоположной тенденции — цены на «черное золото» обвалились. Большинство аналитиков считают, что основную роль в падение цен сыграл мировой финансовой кризис. Только он или есть и другие факторы, которые привели к этой ситуации?
Финансовый кризис сыграл роль катализатора процесса. Однако резкий обвал произошел бы все равно, поскольку за последние годы в мировой экономике накопилась слишком большая психологическая усталость от неоправданно высоких цен на сырьевые товары. Остановка спекулятивного бега была необходима хотя бы на несколько лет.
Сам механизм влияния кризиса достаточно прост: крупномасштабный дефицит ликвидности в финансовой сфере привел сначала к обвалу фондовых рынков, которые неизбежно потянули за собой и товарные. За последние месяцы существенно снизились цены почти на все сырьевые товары, включая металлы, зерно или, например, уголь. То есть произошло общее падение товарных рынков.
Кроме того, инвесторы быстро осознали реальность угрозы рецессии в мировой экономике, что почти автоматически означает (по современным представлениям) временное падение спроса или хотя бы темпов роста спроса на энергоресурсы, особенно со стороны США, ЕС, Японии и Китая. В целом, впервые за последние 25 лет, с 1983 г., мировую экономику ожидает стагнация спроса на нефть.
Наконец, приход к власти в США демократической администрации Барака Обамы несколько охладил накал ближневосточных проблем, особенно ситуацию вокруг Ирана, которая была основным раздражающим фактором для инвесторов все последние годы.
Как следствие всех описанных факторов, нефтяной рынок стал намного менее интересным для спекуляций и при этом слишком рискованным, что и привело к массовому оттоку капитала, обвалившему котировки.
В ежегодном отчете МЕА „World Energy Outlook», который опубликован недавно, говорится, что «эра дешевой нефти закончилось». Председатель правления «Газпрома» Алексей Миллер тоже с уверенности утверждает, что времена дешевых углеводородов прошли. Это так на самом деле? Какое влияние окажут низкие цены на нефть на освоение новых месторождений, которые становится все более технически сложным делом? Грозит ли миру дефицит «черного золота» в ближайшие годы?
После окончания кризиса цены опять начнут расти под воздействием все того же спекулятивного капитала. Однако прогнозировать сейчас точную динамику котировок в 2010-е гг. физически невозможно, поскольку для этого пришлось бы предсказать все войны, ураганы и тысячи иных субъективных факторов, влияющих на спонтанные решения инвесторов. Но, в целом, цены на нефть в 2010-е гг. действительно будут высокими.
В связи с этим особого беспокойства по поводу освоения новых месторождений после 2011 г. у нас нет. Другое дело, насколько сильно ударит по инвестиционным планам компаний экономический кризис. Ведь проблема не только в относительно низких ценах на нефть, которые больше не создают сверхдоходов. Не меньшие сложности вызывает сжимание спроса, распад сложившихся товарных цепочек и, что самое важное, удорожание и дефицит заемных средств, которые играют значительную роль в поддержании текущего платежного баланса большинства нефтегазовых компаний.
В итоге 2009, 2010, а возможно, и 2011 г. будут сложными для нефтяной и газовой отраслей во всех странах, за исключением, пожалуй, лишь ряда аравийских государств, накопивших в 2000-е гг. огромные золотовалютные резервы и не имеющих такой сложной экономики и большого населения, как, например, Россия.
Вместе с тем говорить об угрозе дефицита нефти или природного газа пока нет оснований. Во-первых, сокращение инвестиций в нефтедобычу в период кризиса будет сопровождаться стагнацией спроса, то есть установится некоторое естественное равновесие. Во-вторых, цены на нефть остаются все-таки достаточно высокими для того, чтобы компании сохраняли возможность финансировать ключевые проекты. Под сокращение пойдет, в основном, то, что можно отложить без критической угрозы для производства. Наконец, в странах ОПЕК остается профицит нефтедобывающих мощностей, которых вполне должно хватить в случае всплеска спроса на нефть в первые годы после кризиса.
С природным газом в данном случае ситуация более сложная, поскольку мировой рынок газа все еще очень фрагментирован, по сути его вообще нет. И угроза дефицита на конкретном рынке полностью зависит от ситуации с добычей в связанных с ним странах-экспортерах. Для Европы это, прежде всего, Россия, Норвегия, Алжир и Нигерия.
Эксперты ИПЕМ разработали три основных варианта развития событий на нефтяном рынке. Ваши прогнозы, как будет развиваться этот рынок в перспективы на основе этих моделей?
По нашим расчетам, минимальный уровень, до которого могут упасть цены в период кризиса, составит около 30 долл. за барр. (в текущих ценах). Скорее всего, котировки окажутся даже выше. Вместе с тем в период кризиса рост цен выше 80-90 долл. за барр. также крайне маловероятен. В принципе спекулятивные скачки могут зайти за границы 30-90 долл. и дойти, например, до 20 или опять до 140 долл. за барр., но это будет очень кратковременное явление, буквально на несколько торговых дней, за которыми последует возвращение в коридор 30-90 долл. В целом, на время кризиса мы ожидаем, что средние котировки марки Brent составят около 40-60 долл.
Многие, прежде всего на Западе, считают, что экономический рост в России последних восьми лет был неразрывно связан с ростом нефтяных цен. Какое влияние окажет падение этих цен на российскую экономику? Можно ли говорить, что наступил конец «российской нефтяной сказке»?
Резкое падение цен на нефть и природный газ — это, конечно, очень болезненный удар по всей российской экономике. Но все-таки он не сравним с ударом экономического кризиса в целом.
Падение нефтяных цен создает, прежде всего, проблемы для исполнения трехлетнего государственного бюджета 2009-2011 гг., который рассчитывался изначально из уровня цены 88-95 долл. за барр. Однако все-таки 50 долл. за барр. нефти — это не 10 и не 15 долл., как было в 1990-е гг. Кроме того, накоплены большие ресурсы и есть большой простор для сокращения неоправданно раздутых расходов, если на это хватит политической воли.
Во-вторых, сокращение доходов от экспорта нефти (по газу они пока еще растут) привело к существенному уменьшению притока в Россию валюты, что стало одним из факторов сильного давления на рубль, которое отражается в его плавной девальвации к доллару и евро. Но если для населения это очень негативный фактор, то для промышленности, напротив, весьма позитивный.
Что касается российских нефтяных и газовых компаний, которые также страдают от снижения цен, то их положение далеко не критическое. Есть сложности с выполнением больших инвестиционных программ и с текущим платежным балансом, существует также, конечно, проблема огромных долгов, если речь идет о Газпроме или Роснефти, но по сравнению с 1990-ми годами ситуация опять же намного лучше. Хотя и в 1990-е гг. нефтегазовой комплекс России нельзя сказать, чтобы бедствовал.
Для остальной экономики России падение цен на нефть, скорее, даже положительный фактор: падают цены на автомобильное и авиационное топливо, хотя и не так ощутимо, как в США или ЕС. Иными словами, происходит удешевление грузовых и пассажирских перевозок.
Гораздо чувствительнее для российской экономики сегодня общие последствия мирового финансового кризиса: массовый дефицит ликвидности, удорожание иностранных заемных средств, проблемы с кредитованием, инфляция, падение спроса на российские товары и т.д.
Обвал цен на нефть заставляет не только российских чиновников (министр энергетики РФ Сергей Шматко), но и влиятельных капитанов нефтяной промышленности (вице-президент «ЛУКОЙЛа» Леонид Федун) говорить о необходимости сотрудничества со странами ОПЕК — прежде всего, речь идет о совместном сокращении добычи. Считаете ли Вы это возможным, и какие преимущества или недостатки Вы видите для России при реализации этой идеи?
У России и ОПЕК сейчас сходные интересы: удержать цены на нефть от дальнейшего падения, а по возможности и вернуть их на уровень 70-90 долл. за барр. Однако каких-то реальных механизмов для сотрудничества у нас нет. Добыча нефти в России, после того, как она прошла свой пик в 2007 г., итак начала сокращаться без всяких правительственных решений. В случае падения спроса в Европе (а это наш ключевой рынок сбыта), добычу в любом случае придется урезать. При этом на рынок США, который и делает погоду в динамике мировых котировок, мы практически ничего не поставляет, а потому и влияния на него не оказываем. В Китай же мы не можем прекратить экспорт, поскольку он закреплен межгосударственными соглашениями. В итоге, чтобы целенаправленное снижение добычи нефти в России оказало какое-то серьезное влияние на мировой рынок, оно должно составить, вероятно, не меньше 10-20% (1-2 млн барр./день), на что Россия, естественно, в том числе и по технологическим причинам, пойти не может.
Болгарское информационное агентство БГНЕС
4 декабря 2008 года
Инновации в области цеолитного катализа
Ирина ИВАНОВАПрофессор, д. х. н., главный научный сотрудник, Московский государственный университет имени М. В. Ломоносова, химический факультет
e-mail: [email protected]Ольга ПОНОМАРЕВА
Ведущий научный сотрудник, к. х. н., Московский государственный университет имени М. В. Ломоносова, химический факультет
e-mail: [email protected]Егор АНДРИАКО
Младший научный сотрудник,
Институт нефтехимического синтеза им. А. В. Топчиева РАН
e-mail: [email protected]Николай НЕСТЕРЕНКО
Руководитель программы НИОКР по конверсии природного газа в компании «Тоталь» и лаборатории «Наноклеанэнержи» в национальной инженерной школе
г. Кана (Франция), к. х. н.
e-mail: [email protected]
Цеолитные материалы играют важнейшую роль в нефтепереработке и нефтегазохимии. На фоне новых экологических стандартов, необходимости интеграции процессов нефтепереработки и нефтехимии, возрастающей роли экономики «замкнутого цикла» и водородной энергетики, их вклад будет только увеличиваться. Сегодня эти материалы имеют первостепенное значение для большинства ключевых каталитических процессов нефтегазохимии. Они используются в осушке, очистке, поглощении СО2 из газов горения и позволяют снизить энергозатраты на разделение и очистку и избежать выбросов в атмосферу. Декарбонизация промышленности, необходимость в переработке отходов и пластиков, а также электрификация отрасли, поставят новые задачи перед нефтегазохимией, в которых цеолиты окажутся в центре внимания. Развитие катализаторов и производство адсорбентов на основе цеолитов будет являться приоритетным направлением для жизнеспособности отрасли в последующие 30 лет и сыграет ключевую роль в достижении нулевого уровня выбросов к 2050 году.
Технологии нефтепереработки и нефтегазохимии на основе цеолитных катализаторов
Революционный прорыв в нефтехимии и нефтепереработке, который произошел в середине XX века и привел к существенному повышению эффективности, был связан с внедрением цеолитных катализаторов и созданием современных инновационных технологий на их основе. В настоящее время цеолиты как компоненты катализаторов применяются более чем в половине современных процессов нефтегазохимии.
Цеолиты представляют собой микропористые кристаллические алюмосиликаты с варьируемым составом и размером пор, сравнимым с размерами нефтехимичесих продуктов (0,3–1,2 нм). Мировое производство синтетических цеолитов в 2020 году составило 2200 тыс. тонн, из них 18 % используют в качестве компонентов катализаторов [1]. Основными преимуществами цеолитсодержащих катализаторов являются экологичность, химическая инертность, высокая химическая и термическая стабильность, широкая вариабельность структур, форм-селективность, возможность изменения свойств путем модифицирования, длительность работы, технологичность использования, а также регенерируемость, т. е. восстановление активности путем отжига кокса.
В нефтеперерабатывающей промышленности цеолиты используют в процессах каталитического крекинга (процесс FCC, цеолиты FAU и MFI) и гидрокрекинга (цеолиты FAU), гидроизомеризации С5-С6 парафинов (MOR), гидродеароматизации (FAU, MFI), депарафинизации и изодепарафинизации топлив и масел (MFI, TON, AEL) (рис. 1).
На базе цеолитсодержащих катализаторов созданы технологии изомеризации олефинов (FER) и их олигомеризации (MFI, MTT). В последние годы разработаны и внедрены три технологии твердофазного алкилиривания на цеолитах: Alkyclean® (ABB Lummus-Albemarle), Eurofuel® (Lurgi-Süd Chemie), ExSact ® (Excelus).
В нефтехимии и органическом синтезе цеолитные катализаторы применяются в процессах алкилирования бензола олефинами, ведущих к получению кумола и этилбензола, важных прекурсоров для полимерной промышленности, диспропорционирования толуола (MWW, MOR, MFI) и изомеризации С8 фракции алкилароматических углеводородов (EUO, MOR, MFI), ведущих к получению ксилолов, в процессе окисления пропилена в пропиленоксид (Ti-MFI), а также в ряде других (рис. 2). Замена цеолитсодержащими катализаторами жидких кислот Фриделя – Крафтса или фосфорной кислоты на кизельгуре, на которых проводили, а в большинстве стран, в том числе и в России, до сих пор проводят алкилирование, позволило решить целый ряд экологических, экономических и технологических проблем, связанных с низкой селективностью, коррозией оборудования, загрязнением окружающей среды, большим объемом сточных вод.
В области газохимии актуальной задачей сегодня является разработка цеолитсодержащих катализаторов целевой переработки алканов, которые позволят заменить пиролитические процессы переработки газа каталитическими, а также создание новых технологий монетизации газового сырья и получения на его основе ценных продуктов для промышленности. В настоящее время в газохимии на базе цеолитов MFI и SAPO‑34 внедрены процессы получения из природного газа через метанол/диметиловый эфир высокооктанового бензина (Mobil), этилена и пропилена (UOP, Lurgi, DCIP, Sinopec, JGC, ExxonMobil), изомеризации бутена в изобутилен на FER, и др. [2]
Состояние дел в России
В РФ, в отличие от других стран с высоким уровнем добычи нефти и газа, доля основных вторичных процессов переработки нефти от мощности первичной переработки в среднем составляет около 50 %. При этом общий объем процессов вторичной переработки нефти в России в разы меньше, чем в ведущих странах мира [3]. Решение проблем развития российской нефтегазохимии заложено в Плане развития газо- и нефтехимии на период до 2030 года, утвержденным приказом Минэнерго РФ 01.03.2012 г. Ключевым подходом для выполнения поставленных перед отраслью задач является внедрение новых и усовершенствование существующих технологий, в первую очередь, основанных на применении экологически безопасных твердых цеолитсодержащих катализаторов.
До недавнего времени инновационное развитие отечественной нефтегазохимии строилось на приобретении готовых технологий за рубежом, основанных на импортных катализаторах. В последние годы ситуация несколько изменилась.
В области нефтепереработки были разработаны, внедрены или находятся на стадии внедрения отечественные катализаторы крекинга на основе цеолитов FAU и MFI и гидрокрекинга (FAU), ведущие к получению бензина и дизельного топлива. Основными производителями этих катализаторов в России являются «Ишимбайский специализированный химический завод катализаторов» (10 тыс. т в год при мощности 20 тыс. т в год) и «Газпромнефть – Каталитические системы» (проектная мощность производства 15 тыс. т в год) [4]. В 2017 году на Московском НПЗ «Газпром нефти» внедрен катализатор процесса олигомеризации бутан-бутиленовой фракции, разработанный совместно со специалистами компании УНИСИТ, созданной на базе лаборатории кинетики и катализа химического факультета МГУ. Переход на новый катализатор позволил увеличить пробег и выход целевого продукта, сократить количество регенераций. По подсчетам специалистов Московского НПЗ, годовой эффект от использования нового катализатора олигомеризации превысит 200 млн рублей.
Разработан и готовится к внедрению новый катализатор гидродепарафинизации («Газпром нефть», ВНИИ НП), на основе цеолита MFI, модифицированного гидрирующим компонентом. Ведутся работы по созданию новой технологии твердокислотного алкилирования бутан-бутиленовой фракции на основе цеолитсодержащего катализатора (ИНХС РАН, «Газпром нефть»).
В области нефтехимии ведутся работы по внедрению технологий алкилирования и изомеризации ароматических углеводородов, основанных на цеолитных катализаторах. В 2003 г. в ОАО «Салаватнефтеоргсинтез» внедрен отечественный процесс жидкофазного алкилирования бензола этиленом, разработанный ИНХС РАН, «ГрозНИИ» и «Салаватнефтеоргсинтез» (мощность по этилбензолу 230 тыс. т/год). В настоящее время процесс работает на импортном катализаторе EBEMAX‑1 на основе цеолита MWW (Süd Chemie). С 2010 г. на предприятии «СИБУР-Химпром» действует установка алкилирования бензола этиленом по жидкофазной технологии EBMax с использованием в качестве катализатора цеолита MWW (мощность по этилбензолу 220 тыс. т в год). В 2018 году «Уфаоргсинтез» («Роснефть») завершило модернизацию установки по производству кумола по технологии ExxonMobil (170 тыс. тонн в год). Ведутся работы по модернизации производства кумола на «Омском каучуке» ГК «Титан» по технологии ExxonMobil (165 тыс. т в год), а также запланирована модернизация производства кумола на «Казаньоргсинтезе» на заводе «Бисфенол А» по технологии MobilBadger с использованием цеолитного катализатора MWW.
Уровень развития газохимической отрасли в РФ отстает от передовых стран, только 5 % добываемого газа используется как сырье для вторичной переработки [5]. Процессы, основанные на цеолитсодержащих катализаторах, в России пока не внедрены.
Качество производимого в России цеолита FAU соответствует мировым стандартам, но цеолит MFI по своим свойствам значительно уступает своим импортным аналогам. Цеолит MFI в РФ производится, главным образом, под товарными марками ЦВК, ЦВМ и ЦВН по отечественной технологии, разработанной во ВНИИНП еще в 1970–1980 гг. Если основные мировые производители выпускают цеолиты с размерами кристаллов 200–400 нм, то отечественные предприятия поставляют на рынок цеолит MFI с размером кристаллов 2–5 мкм. Как следствие, катализаторы на основе таких цеолитов характеризуются низкой эффективностью, повышенным коксообразованием и высокой скоростью дезактивации.
Источник: vbashkortostane.gazprom.ru
Вышеперечисленные факторы значительно снижают эффективность отечественных цеолитов MFI в качестве компонентов катализатора крекинга и делают невозможным их применение в процессах олигомеризации и депарафинизации. На настоящий момент потребность российского рынка в высококремнистых цеолитах MFI высокого качества составляет более 500 тонн в год для процессов крекинга, олигомеризации, депарафинизизации, ароматизации и др. Что касается цеолитных катализаторов нефтехимических процессов и процессов органического синтеза на основе высококремнистых среднепористых цеолитов ВЕА, MWW, MOR, ведущих к получению важных мономеров для полимерной промышленности – кумола, этилбензола, ксилолов, то их производство в России отсутствует.
Необходимость перехода к новым экологически чистым технологиям, обеспечивающим устойчивое развитие и прогресс нефтеперерабатывающей, нефтехимической и газохимической отраслей, требует разработки отечественных технологий производства цеолитов. Учитывая введение санкций в отношении России, эта проблема становится крайне актуальной.
В 2019 году в рамках программы Фонда содействия развитию малых форм предприятий в научно-технической сфере, на базе лаборатории кинетики и катализа МГУ была создана компания ООО «Цеолитика», целью которой является разработка новых современных технологий синтеза отечественных наноразмерных цеолитов разных структурных типов (MFI, BEA, MEL, MWW и др.) и их производство в промышленных масштабах для замены импортируемых катализаторов на отечественные. Научным коллективом была предложена новая технология парофазной кристаллизации в отсутствие свободной воды, конкурентными преимуществами которой являются высокое качество получаемых наноразмерных цеолитов, а также отсутствие жидких отходов кристаллизации, эффективное использование исходных реагентов и темплата, уменьшение времени кристаллизации, снижение энергозатрат [6].
Источник: himiya-ufa.ru
Переход к новой энергополитике: инновации в области цеолитного катализа
В настоящее время человечество стоит на пороге очередной революции в области нефтепереработки и нефтехимии. Проблемы рационального использования природных ресурсов, а также все возрастающий вред антропогенного воздействия на окружающий мир вынуждают человечество к глобальному пересмотру подхода к своей деятельности. В ближайшие десятилетия будет осуществлен кардинальный пересмотр процессов нефтехимии и нефтепереработки с возрастающим ростом роли цеолитных материалов. Можно выделить пять основных приоритетных направлений:
- Интеграция нефтепереработки и нефтехимии.
- Операционная эффективность и цифровая трансформация (Индустрия 4.0).
- Декарбонизация нефтехимических комплексов и экономика «замкнутого цикла».
- Переход к новым сырьевым источникам, конверсия биомассы.
- Водородная энергетика и ренессанс газохимии.
Предполагается, что цеолитные катализаторы будут играть первостепенную роль в этом переходе.
Интеграция нефтехимии и нефтепереработки
Рост спроса на базовые продукты нефтехимии опережает рост спроса на топливо, поэтому многие предприятия, чтобы сохранить рентабельность, стремятся к созданию крупных нефтехимических комплексов, объединяющих нефтеперерабатывающие заводы и нефтехимические комплексы. Согласно Hydrocarbon Processing’s Construction Boxscore Database, на сегодняшний день в мире существует около 470 крупных проектов по увеличения мощностей нефтехимических комплексов стоимостью 10 миллиардов долларов. На долю России и стран ближнего зарубежья приходится около 55 проектов (12 %). Все большую популярность приобретают мегапроекты по глубокой интеграции НПЗ и НХК. Эти проекты нацелены на увеличение выхода химикатов на НПЗ с традиционных 10–15 % до 45–53 мас.% [7]. Реализация таких проектов в значительной степени зависит от доступности и готовности новых каталитических технологий. Большинство этих технологий опирается на инновации в области цеолитного катализа в процессах крекинга (FCC, гидрокрекинг), в целевой нефтехимии (селективный крекинг олефинов, каталитический крекинг нафты, дегидрокрекинг нафты, разделение линейных и разветвленных углеводородов, обратная изомеризация) и в ароматическом комплексе (новые технологии по получению ксилолов).
Существует пять основных стратегий интеграции с целью увеличения выхода нефтепродуктов на нефтеперерабатывающих комплексах путем введения технологий «целевой нефтехимии». Первая схема включает модернизацию FCC комплекса путем его интеграции с селективным крекингом тяжелых олефинов из бензиновой фракции в пропилен. Эта схема предполагает оптимизацию цеолитного катализатора для установки FCC и ведет к гораздо более высокому выходу этилена и пропилена. Вторая стратегия предполагает введение процесса гидрокрекинга дистиллята в легкие алканы и нафту с последующим пиролизом алканов в олефины и конверсией нафты в ароматические углеводороды. Следующая стратегия основана на «молекулярном рефайнинге» на цеолитах, включающем разделение СУГ на пропан и бутан с последующей их конверсией в пропилен и бутены, а также разделение линейных и разветвленных парафинов в нафте с последующей изомеризацией разветвленных углеводородов в линейные и их пиролизом в этилен и пропилен. В результате этой операции только нормальные парафины подвергаются конверсии в установке пиролиза, что увеличивает выход легких олефинов на 10–15 %. Четвертая стратегия связана с введением процесса метатезиса олефинов, что открывает широкие возможности для увеличения выхода этилена и пропилена. И, наконец, пятая стратегия основана на каталитическом (дегидро-) крекинге нафты, который позволяет значительно повысить выход пропилена и этилена по сравнению с процессом пиролиза. Реализация перечисленных стратегий позволит повысить выход нефтепродуктов с 10–15 % до 30–50 %, что соответствует полностью интегрированному комплексу.
Следующим этапом эволюции будет прямая конверсия нефти в продукты нефтехимии путем крекинга. Этот вариант ведет к снижению производственных затрат и выбросов СО2 на тонну продукта. На настоящий момент существует как минимум три стратегии для осуществления этого этапа. Они основаны на процессах FCC, гидрокрекинга и пиролиза. Выбор схемы зависит от вида сырья и типа нефтепродуктов, которые завод должен производить. Во всех трех случаях, инновации в области цеолитного катализа будут играть основополагающую роль.
Операционная эффективность и цифровая трансформация (Индустрия 4.0)
Основой операционной эффективности производства является надежность и детальное знание процесса и катализатора. Цеолитные катализаторы более 60 лет используются в промышленности и зарекомендовали себя как надежные, безопасные и эффективные. Однако разработка новых технологий на основе цеолитов занимает длительное время, в среднем от 10 до 15 лет, что не совместимо с ростом спроса на эти технологии. Увеличение скорости внедрения новых разработок может быть достигнуто путем использования моделирования, информатики материалов и искусственного интеллекта. Необходимым условием внедрения технологий цифровой трансформации в этой области является создание библиотек цеолитных материалов и катализаторов, подготовка специалистов и поддержка научных школ в области цеолитного катализа.
Декарбонизация нефтехимических комплексов и экономика «замкнутого цикла»
В соответствии с Парижским соглашением по климату от 2015 года, главным врагом климата объявлены парниковые газы, в основном диоксид углерода, а значит, косвенно, и источники его образования – нефть, газ, уголь. В мировой практике по инициативе ЕС вводится система отчетности по так называемому «углеродному следу», другими словами при производстве любого вида товаров и услуг будет оцениваться эмиссия парниковых газов. Правительства практически всех стран мира вводят углеродный налог – плату за выбросы эквивалента углекислого газа в атмосферу. По оценке российской академии наук, финансовые потери отечественных экспортеров могут достичь 3–4 млрд евро в год.
Таким образом, для российской нефтегазохимии необходимо пересмотреть сложившиеся реалии экспортно-сырьевого развития. Основой развития должен стать выпуск высокотехнологичной, высококачественной продукции более высоких переделов на базе ресурсо- и трудосберегающих экологически чистых технологий.
В первую очередь потребуется внедрение технологий для улавливания углекислого газа, его хранения и переработки. Цеолитные технологии будут играть первостепенную роль в этом отношении [8]. Прежде всего, цеолитные материалы могут быть использованы для улавливания СО2 из газов горения при низком парциальном давлении компонентов и его очистки для последующей транспортировки. Использование цеолитов в процессах разделения позволит значительно снизить потребление энергии и, соответственно, выбросы антропогенных газов.
Помимо этого, важной задачей станет утилизация СО2 путем его конверсии в полезные продукты. В этом отношении перспективными являются процессы «замкнутого цикла». В настоящее время рассматриваются 2 таких цикла, которые взаимосвязаны между собой (рис. 3). Наиболее приемлемым решением для утилизации СО2 будет его конверсия в метанол. Метанол может найти большое количество применений на нефтеперерабатывающем комплексе, но одним из наиболее перспективных решений будет его конверсия в олефины, ароматические углеводороды и в топлива (процессы MTO, MTG). Процесс получения метанола потребует использования больших количеств водорода, который также может быть получен на основе цеолитных технологий (пп 3.5).
Процессы конверсии метанола в олефины и ароматические углеводороды весьма перспективны в связи со значительным ростом спроса на пластики, получаемые из этих мономеров. Получение пластиков из метанола будет решать задачу экономики «замкнутого цикла», которая является одной из наиболее важных задач для нефтехимии сегодня. Отходы от использования пластиков все больше рассматриваются как один из основных источников загрязнений и обеспечение их эффективной переработки является необходимым для существования отрасли. Наиболее оптимальным направлением такой переработки будет термический пиролиз, продукты которого будут перерабатываться на интегрированных нефтехимических комплексах, на установках FCC и гидрокрегинга на цеолитных катализаторах, давая в конечном результате олефины и ароматические углеводороды, необходимые для синтеза пластиков. Цеолиты, таким образом, будут продолжать играть первостепенную роль в процессах «замкнутого цикла».
Еще одним аспектом экономики «замкнутого цикла» является электрификация конверсии углеводородов. В настоящее время большинство печей, компрессоров и генераторов водяного пара на НПЗ используют углеводороды в качестве топлива. В результате сжигания этих продуктов образуются СО2‑содержащие газы. Абсорбция аминами не эффективна для этих типов потоков и улавливание углекислого газа из них стоит очень дорого. Поэтому одной из современных тенденций является замена топливных нагревательных элементов на электрические, в которых энергия будет подаваться в объем реактора. Другими словами, не стенки реактора будут подвергаться нагреву, а катализатор. Это также значительно упростит регенерацию и снизит инертность систем. Новое поколение катализаторов на основе цеолитов должно быть разработано для этих типов установок.
Переход к новым сырьевым источникам, конверсия биомассы
Растущий спрос на биотопливо обусловлен несколькими ключевыми причинами, первая из которых заключается в том, что биоресурсы являются возобновляемыми и нейтральными по отношению к CO2 в отличие от ископаемого топлива. Использование биомассы для производства топлива и химикатов может привести к частичному замещению ископаемых ресурсов возобновляемыми источниками. Многие государства в последнее десятилетие выделяют большие ресурсы для создания технологий для переработки биомассы [9].
Биомасса, как и нефть, состоит из высокомолекулярных компонентов. Основными источниками биомассы являются лигноцеллюлоза и липиды, которые содержат триглицериды из растительных животных жиров и микроводорослей. Из них наиболее распространенной является лигноцеллюлозная биомасса, состоящая из целлюлозы, гемицеллюлозы и лигнина. Кроме того, существует меньшая группа белковых фракций животного или растительного происхождения. Схема переработки биомассы приведена на рис. 4.
Основная цель валоризации биомассы состоит в уменьшении молекулярной массы. По аналогии с процессами конверсии нефти здесь могут быть использованы процессы каталитического крекинга на цеолитных катализаторах. Жиры и масла, имеющие длинные линейные углеводородные цепи, структурно наиболее близки к нефтехимическим углеводородам, поэтому в данных процессах цеолиты используются с большим успехом. Однако следует ожидать, что механизмы превращения громоздких субстратов биомассы, содержащих большое количество кислорода, более сложные и требуют создания новых каталитических систем [10]. Рядом компаний разработаны процессы прямого крекинга в установках типа FCC сухой биомассы. Ко-процессинг на установках FCC или гидрокрекинга нефтяного сырья с биосырьем, прошедшим этапы частичного гидрирования или термического пиролиза, является оптимальным решением во многих случаях. Это также позволяет избежать дополнительных инвестиций и значительных изменений в операционных схемах.
Другим интересным вариантом является конверсия спиртов. Этанол и бутанол в настоящее время в больших количествах производятся из возобновляемых источников. Помимо их использования в качестве биотоплива, конверсия биоэтанола и биобутанола в ценные химические вещества становится все более привлекательной. Перспективные технологии включают конверсию этанола в этилен, пропилен и бутадиен, а также конверсию бутанола в бутены, легкие олефины и ароматические углеводороды. Для этих процессов разработаны эффективные цеолитные катализаторы.
Водородная энергетика и ренессанс газохимии
Водород является высокоэнергетическим продуктом и может быть использован как для внутренних нужд нефтехимических комплексов, так и для конверсии CO2. На сегодняшний день основным способом производства водорода является паровая конверсия. Однако этот способ приводит к выделению 9,3 тонн СО2 на тонну водорода, который требует улавливания и соответствующей инфраструктуры для этого (рис. 5). Альтернативная технология, основанная на электролизе воды, поглощает в 5 раз большее количество энергии на тонну водорода. В связи с этим, прямая декарбонизация природного газа в углерод, олефины или ароматические углеводороды приобретает все большее значение для водородной энергетики. Первый процесс имеет серьезный недостаток, так как приводит к образованию большого количества кокса, который не имеет рынка сбыта. Поэтому, процессы превращения метана в водород и олефины или ароматику гораздо более перспективны. Эти процессы могут эффективно протекать на цеолитах. Многие из этих технологий находятся в стадии разработки и будут играть значительную роль для нефтегазохимии.
Кроме того, все большее значение в будущем будут иметь процессы получения водорода путем ароматизации легких алканов, доля которых увеличится из-за интеграции нефтехимических комплексов. В основе этих процессов также лежат цеолитные катализаторы (процессы CICLAR (UOP), ALPHA(ASAHI), AROMAX(CHEVRON)).
Заключение
Анализ перспектив развития нефтегазохимической отрасли в области цеолитного катализа показывает, что первоочередной задачей здесь является разработка и внедрение отечественной технологии синтеза наноразмерных высококремнистых цеолитов структурных типов MFI, BEA, MWW и MOR, необходимых для обеспечения действующих нефтехимических производств.
Кроме того, актуальной задачей представляется создание новых цеолитных катализаторов и технологий для обеспечения интеграции НПЗ и НХК; создания прямых процессов конверсии нефти в ценные продукты нефтехимии; реализации процессов «замкнутого цикла» – конверсии CO2, отходов и пластиков в мономеры и топлива; осуществление конверсии биомассы в топлива и ценные химические продукты, а также конверсии метана в водород и олефины или ароматические углеводороды. Реализация этих задач позволит осуществить переход к новой энергетической политике.
Центральная роль ферментов как биологических катализаторов — клетка
Фундаментальная задача белков — действовать как ферменты — катализаторы, которые увеличивают скорость практически всех химических реакций внутри клеток. Хотя РНК способны катализировать некоторые реакции, большинство биологических реакций катализируются белками. В отсутствие ферментативного катализа большинство биохимических реакций протекают настолько медленно, что они не могли бы происходить в мягких условиях температуры и давления, совместимых с жизнью.Ферменты ускоряют скорость таких реакций более чем в миллион раз, поэтому реакции, на которые в отсутствие катализа ушли бы годы, могут происходить за доли секунды, если их катализирует соответствующий фермент. Клетки содержат тысячи различных ферментов, и их активность определяет, какая из многих возможных химических реакций действительно происходит внутри клетки.
Каталитическая активность ферментов
Как и все другие катализаторы, ферменты характеризуются двумя основными свойствами.Во-первых, они увеличивают скорость химических реакций, при этом сами по себе реакция не поглощается и не изменяется навсегда. Во-вторых, они увеличивают скорость реакции без изменения химического равновесия между реагентами и продуктами.
Эти принципы ферментативного катализа проиллюстрированы в следующем примере, в котором молекула, на которую воздействует фермент (называемая субстратом [ S ]), превращается в продукт ( P ) в результате реакции.В отсутствие фермента реакция может быть записана следующим образом:
Химическое равновесие между S и P определяется законами термодинамики (как обсуждается далее в следующем разделе этой главы) и представлено по соотношению скоростей прямой и обратной реакции ( S → P и P → S соответственно). В присутствии соответствующего фермента превращение S в P ускоряется, но равновесие между S и P не изменяется.Следовательно, фермент должен одинаково ускорять как прямую, так и обратную реакции. Реакция может быть записана следующим образом:
Обратите внимание, что фермент ( E ) не изменяется в результате реакции, поэтому химическое равновесие остается неизменным, определяемым исключительно термодинамическими свойствами S и P .
Влияние фермента на такую реакцию лучше всего иллюстрируется изменениями энергии, которые должны произойти во время превращения S в P ().Равновесие реакции определяется конечными энергетическими состояниями S и P , на которые не влияет ферментативный катализ. Однако для того, чтобы реакция продолжилась, подложку необходимо сначала преобразовать в состояние с более высокой энергией, называемое переходным состоянием . Энергия, необходимая для достижения переходного состояния (энергия активации), представляет собой барьер для развития реакции, ограничивая скорость реакции. Ферменты (и другие катализаторы) действуют за счет снижения энергии активации, тем самым увеличивая скорость реакции.Повышенная скорость одинакова как в прямом, так и в обратном направлениях, поскольку оба должны проходить через одно и то же переходное состояние.
Рис. 2.22
Энергетические диаграммы для катализированных и некаталитических реакций. Проиллюстрированная реакция представляет собой простое преобразование субстрата S в продукт P Поскольку конечное энергетическое состояние P ниже, чем у S , реакция протекает слева направо. Для (подробнее …)
Каталитическая активность ферментов включает связывание их субстратов с образованием комплекса фермент-субстрат ( ES ).Субстрат связывается с определенной областью фермента, называемой активным центром. Связавшись с активным центром, субстрат превращается в продукт реакции, который затем высвобождается из фермента. Таким образом, реакция, катализируемая ферментами, может быть записана следующим образом:
Обратите внимание, что E остается неизменным с обеих сторон уравнения, поэтому равновесие не нарушается. Однако фермент обеспечивает поверхность, на которой реакции, превращающие S в P , могут протекать более легко.Это результат взаимодействий между ферментом и субстратом, которые снижают энергию активации и способствуют образованию переходного состояния.
Механизмы ферментативного катализа
Связывание субстрата с активным центром фермента является очень специфическим взаимодействием. Активные сайты — это щели или бороздки на поверхности фермента, обычно состоящие из аминокислот из разных частей полипептидной цепи, которые собраны вместе в третичной структуре свернутого белка.Субстраты изначально связываются с активным центром за счет нековалентных взаимодействий, включая водородные связи, ионные связи и гидрофобные взаимодействия. Как только субстрат связывается с активным центром фермента, несколько механизмов могут ускорить его превращение в продукт реакции.
Хотя простой пример, обсужденный в предыдущем разделе, включает только одну молекулу субстрата, большинство биохимических реакций включают взаимодействия между двумя или более различными субстратами. Например, образование пептидной связи включает соединение двух аминокислот.Для таких реакций связывание двух или более субстратов с активным центром в правильном положении и ориентации ускоряет реакцию (). Фермент обеспечивает матрицу, на которой реагенты собираются вместе и должным образом ориентируются, чтобы способствовать образованию переходного состояния, в котором они взаимодействуют.
Рис. 2.23
Ферментативный катализ реакции между двумя субстратами. Фермент обеспечивает матрицу, на которой два субстрата сводятся вместе в правильном положении и ориентации для взаимодействия друг с другом.
Ферменты ускоряют реакции, также изменяя конформацию своих субстратов, чтобы приблизиться к конформации переходного состояния. Простейшей моделью взаимодействия фермент-субстрат является модель lock-and-key , в которой субстрат точно входит в активный центр (). Однако во многих случаях конфигурации как фермента, так и субстрата модифицируются связыванием субстрата — процесс, называемый индуцированной подгонкой . В таких случаях конформация субстрата изменяется так, что она больше напоминает конформацию переходного состояния.Напряжение, вызванное таким искажением подложки, может дополнительно облегчить ее переход в переходное состояние за счет ослабления критических связей. Более того, переходное состояние стабилизируется за счет его прочного связывания с ферментом, тем самым снижая требуемую энергию активации.
Рисунок 2.24
Модели взаимодействия фермент-субстрат. (A) В модели с замком и ключом субстрат точно входит в активный центр фермента. (B) В модели индуцированной подгонки связывание субстрата искажает конформации как субстрата, так и фермента.Это искажение (подробнее …)
Помимо объединения нескольких субстратов и искажения конформации субстратов для достижения переходного состояния, многие ферменты непосредственно участвуют в каталитическом процессе. В таких случаях боковые цепи конкретных аминокислот в активном центре могут реагировать с субстратом и образовывать связи с промежуточными продуктами реакции. Кислые и основные аминокислоты часто участвуют в этих каталитических механизмах, как показано в следующем обсуждении химотрипсина как примера ферментативного катализа.
Химотрипсин является членом семейства ферментов (сериновых протеаз), которые переваривают белки, катализируя гидролиз пептидных связей. Реакцию можно записать следующим образом:
Различные члены семейства сериновых протеаз (включая химотрипсин, трипсин, эластазу и тромбин) имеют различные субстратные специфичности; они предпочтительно расщепляют пептидные связи, прилегающие к различным аминокислотам. Например, в то время как химотрипсин расщепляет связи, соседние с гидрофобными аминокислотами, такими как триптофан и фенилаланин, трипсин расщепляет связи рядом с основными аминокислотами, такими как лизин и аргинин.Однако все сериновые протеазы похожи по структуре и используют один и тот же механизм катализа. Активные центры этих ферментов содержат три важные аминокислоты — серин, гистидин и аспартат, которые управляют гидролизом пептидной связи. Действительно, эти ферменты называются сериновыми протеазами из-за центральной роли серинового остатка.
Субстраты связываются с сериновыми протеазами путем встраивания аминокислоты, прилегающей к сайту расщепления, в карман на активном сайте фермента ().Природа этого кармана определяет субстратную специфичность различных членов семейства сериновых протеаз. Например, связывающий карман химотрипсина содержит гидрофобные аминокислоты, которые взаимодействуют с гидрофобными боковыми цепями его предпочтительных субстратов. Напротив, связывающий карман трипсина содержит отрицательно заряженную кислотную аминокислоту (аспартат), которая способна образовывать ионную связь с остатками лизина или аргинина своих субстратов.
Рисунок 2.25
Связывание субстрата сериновыми протеазами.Аминокислота, примыкающая к расщепляемой пептидной связи, вставляется в карман в активном центре фермента. В химотрипсине карман связывает гидрофобные аминокислоты; связывающий карман трипсина содержит (подробнее …)
Субстрат связывается с расщепляемой пептидной связью рядом с серином активного сайта (). Затем протон этого серина переносится на гистидин в активном центре. Конформация активного центра способствует переносу протона, поскольку гистидин взаимодействует с отрицательно заряженным остатком аспартата.Серин реагирует с субстратом, образуя тетраэдрическое переходное состояние. Затем пептидная связь расщепляется, и фермент высвобождает С-концевую часть субстрата. Однако N-концевой пептид остается связанным с серином. Эта ситуация разрешается, когда молекула воды (второй субстрат) входит в активный центр и меняет предыдущие реакции. Протон молекулы воды переносится на гистидин, а его гидроксильная группа переносится на пептид, образуя второе тетраэдрическое переходное состояние.Затем протон переносится от гистидина обратно к серину, и пептид высвобождается из фермента, завершая реакцию.
Рисунок 2.26
Каталитический механизм химотрипсина. Три аминокислоты в активном центре (Ser-195, His-57 и Asp-102) играют решающую роль в катализе.
Этот пример иллюстрирует несколько особенностей ферментативного катализа; специфичность взаимодействий фермент-субстрат, расположение различных молекул субстрата в активном центре и участие остатков активного сайта в формировании и стабилизации переходного состояния.Хотя тысячи ферментов в клетках катализируют множество различных типов химических реакций, к их работе применимы одни и те же основные принципы.
Коферменты
Помимо связывания своих субстратов, активные центры многих ферментов связывают другие небольшие молекулы, которые участвуют в катализе. Протезные группы — это небольшие молекулы, связанные с белками, в которых они играют важнейшие функциональные роли. Например, кислород, переносимый миоглобином и гемоглобином, связан с гемом, простетической группой этих белков.Во многих случаях ионы металлов (например, цинка или железа) связаны с ферментами и играют центральную роль в каталитическом процессе. Кроме того, различные низкомолекулярные органические молекулы участвуют в определенных типах ферментативных реакций. Эти молекулы называются коферментами, потому что они работают вместе с ферментами, увеличивая скорость реакции. В отличие от субстратов, коферменты не изменяются необратимо в результате реакций, в которых они участвуют. Скорее они перерабатываются и могут участвовать во множестве ферментативных реакций.
Коферменты служат переносчиками нескольких типов химических групп. Ярким примером кофермента является никотинамидадениндинуклеотид ( NAD + ), который функционирует как переносчик электронов в реакциях окисления-восстановления (). NAD + может принимать ион водорода (H + ) и два электрона (e — ) от одной подложки, образуя NADH. Затем НАДН может отдавать эти электроны второй подложке, повторно образуя НАД + .Таким образом, NAD + переносит электроны от первой подложки (которая окисляется) ко второй (которая восстанавливается).
Рисунок 2.27
Роль NAD + в окислительно-восстановительных реакциях. (A) Никотинамидадениндинуклеотид (NAD + ) действует как переносчик электронов в окислительно-восстановительных реакциях, принимая электроны (e —) с образованием NADH. (B) Например, NAD + может принимать электроны от одного субстрата (подробнее …)
Несколько других коферментов также действуют как переносчики электронов, а третьи участвуют в переносе множества дополнительных химических групп (e .g. карбоксильные группы и ацильные группы; ). Одни и те же коферменты действуют вместе с множеством различных ферментов, катализируя перенос определенных химических групп между широким спектром субстратов. Многие коферменты тесно связаны с витаминами, которые вносят часть или всю структуру кофермента. Витамины не требуются бактериям, таким как E. coli , но они являются необходимыми компонентами рациона человека и других высших животных, которые утратили способность синтезировать эти соединения.
Регуляция активности ферментов
Важной особенностью большинства ферментов является то, что их активность непостоянна, а вместо этого может модулироваться. То есть активность ферментов можно регулировать, чтобы они функционировали надлежащим образом для удовлетворения разнообразных физиологических потребностей, которые могут возникнуть в течение жизни клетки.
Одним из распространенных типов регуляции ферментов является ингибирование с обратной связью, при котором продукт метаболического пути подавляет активность фермента, участвующего в его синтезе.Например, аминокислота изолейцин синтезируется серией реакций, начиная с аминокислоты треонина (). Первый этап этого пути катализируется ферментом треониндезаминазой, который ингибируется изолейцином, конечным продуктом этого пути. Таким образом, достаточное количество изолейцина в клетке ингибирует треониндезаминазу, блокируя дальнейший синтез изолейцина. Если концентрация изолейцина снижается, подавление обратной связи снимается, треониндезаминаза больше не ингибируется и синтезируется дополнительный изолейцин.Регулируя таким образом активность треониндезаминазы, клетка синтезирует необходимое количество изолейцина, но не тратит энергию на синтез большего количества изолейцина, чем необходимо.
Рисунок 2.28
Запрет обратной связи. Первый этап превращения треонина в изолейцин катализируется ферментом треониндезаминазой. Активность этого фермента подавляется изолейцином, конечным продуктом метаболизма.
Ингибирование по обратной связи является одним из примеров аллостерической регуляции, при которой активность фермента контролируется связыванием малых молекул с регуляторными участками фермента ().Термин «аллостерическая регуляция» происходит от того факта, что регуляторные молекулы связываются не с каталитическим сайтом, а с отдельным сайтом на белке ( алло = «другой» и стерический = «сайт»). Связывание регуляторной молекулы изменяет конформацию белка, что, в свою очередь, изменяет форму активного центра и каталитическую активность фермента. В случае треониндезаминазы связывание регуляторной молекулы (изолейцина) подавляет ферментативную активность. В других случаях регуляторные молекулы служат активаторами, стимулируя, а не ингибируя их целевые ферменты.
Рисунок 2.29
Аллостерическая регуляция. В этом примере активность фермента ингибируется связыванием регуляторной молекулы с аллостерическим сайтом. В отсутствие ингибитора субстрат связывается с активным центром фермента, и реакция продолжается. Связывание (подробнее …)
Активность ферментов также может регулироваться их взаимодействием с другими белками и ковалентными модификациями, такими как добавление фосфатных групп к остаткам серина, треонина или тирозина.Фосфорилирование — особенно распространенный механизм регулирования активности ферментов; добавление фосфатных групп либо стимулирует, либо подавляет активность многих различных ферментов (). Например, мышечные клетки реагируют на адреналин (адреналин), расщепляя гликоген на глюкозу, тем самым обеспечивая источник энергии для повышенной мышечной активности. Распад гликогена катализируется ферментом гликогенфосфорилазой, который активируется фосфорилированием в ответ на связывание адреналина с рецептором на поверхности мышечной клетки.Фосфорилирование белков играет центральную роль в контроле не только метаболических реакций, но и многих других клеточных функций, включая рост и дифференцировку клеток.
Рисунок 2.30
Фосфорилирование белков. Некоторые ферменты регулируются добавлением фосфатных групп к ОН-группам боковой цепи серина (как показано здесь), треонина или остатков тирозина. Например, фермент гликогенфосфорилаза, который катализирует преобразование (подробнее …)
Роль катализатора в контроле роста и морфологии одномерных наноструктур SiC
Контроль морфологии одномерных (1D) наноструктур, особенно во время роста полупроводниковых наноструктур с помощью катализатора, является ключом к раскрытию их потенциальных применений.В этой работе мы демонстрируем, что морфологией одномерных наноструктур карбида кремния (SiC) можно управлять, изменяя состав катализатора в процессе микроволнового плазменного химического осаждения из паровой фазы. Выявлено, что силицид железа является основным катализатором роста одномерной наноструктуры SiC. Анализ с помощью просвечивающей электронной микроскопии с высоким разрешением показывает, что стехиометрия силицида железа определяет окончательную морфологию нанопроволоки 1D SiC. Для роста нанопроволок SiC в качестве катализатора используется Fe 5 Si 3 , а для SiC наноигл — Fe 3 Si.Особое соответствие ориентации катализатора на основе силицида железа и нанопроволоки SiC наблюдается впервые при росте наноструктур SiC. Считается, что механизмом различной морфологии наноструктур SiC является различное сопротивление травлению частиц катализатора при плазменном травлении H 2 . На основе вышеупомянутого механизма непрерывное изменение морфологии наноструктур SiC было достигнуто путем управления подачей Si во время роста.
Эта статья в открытом доступе
Подождите, пока мы загрузим ваш контент… Что-то пошло не так. Попробуй снова?Произошла ошибка при установке пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно. Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки своего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались. Чтобы принять файлы cookie с этого сайта, нажмите кнопку «Назад» и примите файлы cookie.
- Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г., браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере.
- Вы установили приложение, которое отслеживает или блокирует установку файлов cookie. Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу. Чтобы предоставить доступ без файлов cookie потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в файле cookie может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта. Например, сайт не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
17.6: Катализаторы и катализ — Chemistry LibreTexts
Цели обучения
Убедитесь, что вы полностью понимаете следующие важные идеи, которые были представлены выше.Особенно важно, чтобы вы знали точное значение всех выделенных зеленым цветом терминов в контексте этой темы.
- Что такое катализаторы и как они работают, изменяя параметры реакции?
- Опишите сходства и различия между тремя основными классами катализаторов.
- Определите физическую сорбцию , и хемосорбцию , и объясните роль последней в инициировании каталитического события.
- В чем смысл и значение Принцип Сабатье ?
- Каковы принципиальные различия между механизмами гетерогенного катализа Langmuir-Hinshelwood и Eley-Rideal ?
- Опишите роль комплекса фермент-субстрат .
- Каким образом особые свойства аминокислот , окружающих активный центр фермента, могут способствовать его каталитическому эффекту?
- Опишите модели действия ферментов с замком и ключом, и , индуцированные подгонкой, .
- Объясните функцию и значение аллостерических сайтов на ферменте.
Это почти похоже на волшебство! Смесь газообразных H 2 и O 2 может сосуществовать неопределенно долго без какой-либо обнаруживаемой реакции, но если платиновую проволоку нагреть, а затем погрузить в газовую смесь, реакция протекает мягко, так как выделяющееся при реакции тепло заставляет проволоку светиться докрасна. Катализаторы играют важную роль в нашей современной индустриальной экономике, в заботе об окружающей среде и во всех биологических процессах.Этот урок познакомит вас с удивительным миром катализаторов и поможет понять, что они из себя представляют и как работают.
Что такое катализаторы?
Катализаторы не влияют на константу равновесия и, следовательно, на равновесный состав. Катализаторы — это вещества, которые ускоряют реакцию, но не потребляются ею и не фигурируют в итоговом уравнении реакции. Также — и это очень важно — катализаторы одинаково влияют на скорость прямого и обратного хода ; это означает, что катализаторы не влияют на константу равновесия и, следовательно, на состав состояния равновесия.Таким образом, катализатор (в данном случае серная кислота) может быть использован для ускорения обратимой реакции, такой как образование сложного эфира или его обратный гидролиз сложного эфира:
Рисунок \ (\ PageIndex {1} \): Реакции, катализируемые кислотойКатализатор не влияет на константу равновесия или направление реакции. Направление можно контролировать, добавляя или удаляя воду (принцип Ле Шателье).
Катализаторы функционируют, позволяя реакции протекать через альтернативный механизм, требующий меньшей энергии активации.Это изменение вызвано особым взаимодействием между катализатором и компонентами реакции. Вы помните, что константа скорости реакции является экспоненциальной функцией энергии активации, поэтому даже небольшое уменьшение \ (E_a \) может дать впечатляющее увеличение скорости.
Катализаторы обеспечивают альтернативные пути реакции
Катализаторы условно делятся на две категории: гомогенные и гетерогенные . Ферменты , природные биологические катализаторы, часто включаются в первую группу, но поскольку они обладают некоторыми свойствами обоих, но проявляют некоторые особые свойства, мы будем рассматривать их здесь как третью категорию.
Некоторые общие примеры катализа
Как сжечь сахарный кубик
При нагревании кусочек сахара (сахароза) плавится при 185 ° C, но не горит. Но если натереть кубик сигаретным пеплом, он сгорит перед расплавлением из-за каталитического действия следов соединений металлов в золе.
Платина как катализатор окисления
Поверхность металлической платины является эффективным катализатором окисления многих паров топлива. Это свойство используется в беспламенных походных печах (слева).Изображение справа показывает светящуюся платиновую проволоку, нагретую за счет медленного горения аммиака на ее поверхности. Однако если окунуть нагретую Pt проволоку в жидкий аммиак , получится миниатюрный взрыв: см. Видео ниже.
Это окисление аммиака кислородом, катализируемое нагретой платиновой проволокой. Кислород барботируется через раствор аммиака, в котором он смешивается с присутствующим газообразным аммиаком. В результате реакции платиновая проволока загорается, а горячая проволока воспламеняет смесь аммиака и кислорода.{–1} \]
В отсутствие примесей эта реакция протекает очень медленно, но различные вещества, начиная от йода, оксидов металлов и следовых количеств металлов, значительно ускоряют реакцию, в некоторых случаях почти взрывоопасно из-за быстрого выделения тепла. Наиболее эффективным катализатором из всех является фермент каталаза , присутствующий в крови и внутриклеточных жидкостях; добавление капли крови к раствору 30% перекиси водорода вызывает бурную реакцию.
Быстрое высвобождение O 2 может привести к эффектной пены для ванны, если добавить немного мыла.[Эта же реакция использовалась для запуска гоночного автомобиля!
Иодид калия эффективно катализирует осаждение H 2 O 2 . В этом коротком видео показано, что происходит при объединении цветного мыла, H 2 O 2 и KI. Но «не пробуйте это у себя дома»!
Каждый тип катализатора способствует разному пути со своей собственной энергией активации. Поскольку скорость является экспоненциальной функцией от E до (уравнение Аррениуса), даже относительно небольшие различия в E и могут иметь драматическое влияние на скорость реакции.Обратите особое внимание на значения для каталазы; химик по-прежнему остается непрофессиональным любителем по сравнению с тем, что Природа может сделать с помощью естественного отбора!
| катализатор | Ea | относительный коэффициент |
|---|---|---|
| без катализатора | 75 | 1 |
| иодид-ион | 56 | 2140 |
| платина коллоидная | 50 | 24 000 |
| каталаза (фермент) | 21 | 2 900 000 000 |
Как выражается каталитическая активность
Изменения константы скорости или энергии активации — очевидные способы измерения эффективности катализатора.Но вошли в употребление два других термина, которые имеют особое значение в промышленных приложениях.
Оборотный номер
Число оборотов (TON) — это среднее количество циклов, которое катализатор может пройти до того, как его характеристики ухудшатся (см.). Зарегистрированные значения TON для обычных промышленных катализаторов охватывают очень широкий диапазон от 10 до 10 5 , что приближается к пределам диффузионного переноса.
Частота оборота
Этот термин, который первоначально применялся к реакциям, катализируемым ферментами, вошел в более широкое употребление.Это просто количество раз, когда общая каталитическая реакция происходит на катализатор (или на активный центр на ферменте или гетерогенном катализаторе) в единицу времени: определяется как
Число активных центров S на гетерогенном катализаторе часто трудно оценить, поэтому его часто заменяют общей площадью открытого катализатора, которую обычно можно измерить экспериментально. TOF для гетерогенных реакций обычно находится в диапазоне от 10 –2 до 10 2 с –1 .
Гомогенный катализ
Как следует из названия, гомогенные катализаторы присутствуют в той же фазе (газ или жидкий раствор), что и реагенты. Гомогенные катализаторы обычно вступают непосредственно в химическую реакцию (путем образования нового соединения или комплекса с реагентом), но высвобождаются в исходной форме после завершения реакции, так что они не появляются в итоговом уравнении реакции.
Катализируемая йодом
Цис-транс ИзомеризацияЕсли вы не изучаете курс органической химии, на котором ваш инструктор указывает иное, не пытайтесь запоминать эти механизмы .Они представлены здесь с целью убедить вас, что катализ — это не черная магия, и познакомить вас с некоторыми особенностями каталитических механизмов. Вам должно быть достаточно просто убедить себя, что отдельные шаги имеют химический смысл.
Как вы помните, цис-транс-изомерия возможна, когда атомы, связанные с каждым из двух углеродов с двойной связью, могут находиться на одной ( цис ) или противоположных сторонах связи ( транс ).Это отражает тот факт, что вращение вокруг двойной связи невозможно.
Превращение алкена между его формами цис — и транс может происходить только в том случае, если двойная связь временно разорвана, что позволяет двум концам вращаться. Процессы, расщепляющие ковалентные связи, имеют высокую энергию активации, поэтому реакции цис-транс-изомеризации имеют тенденцию быть медленными даже при высоких температурах. Йод является одним из нескольких катализаторов, которые значительно ускоряют этот процесс, поэтому изомеризация бутена служит хорошим вводным примером гомогенного катализа.
Считается, что механизм реакции, катализируемой йодом, включает атаку атомов йода (образованных равновесием диссоциации на одном из атомов углерода на этапе:
За время своего недолгого существования активированный комплекс в свободном состоянии может вращаться вокруг связи C — C, так что, когда он разлагается с высвобождением йода (), часть восстановленного бутена будет в его форме транс . Наконец, атом йода рекомбинирует в дийод.Поскольку процессы и нейтрализуются, йод не появляется в общем уравнении реакции — требование для настоящего катализатора.
Кислотно-основной катализ
Многие реакции катализируются присутствием кислоты или основания; во многих случаях и кислоты, и основания катализируют одну и ту же реакцию. Как и следовало ожидать, механизм включает добавление или удаление протона, превращая реагент в более кинетически лабильную форму. Простым примером является добавление йода к пропанону
.I 2 + (CH 3 ) 2 –C = O → (CH 2 I) (CH 3 ) –C = O
Механизм кислотно-катализируемого процесса включает несколько этапов.Роль кислоты заключается в обеспечении протона, который присоединяется к карбонильному кислороду, образуя нестабильный ион оксония. Последний быстро перестраивается в енол (то есть углерод, связанный как с двойной связью ( ен ), так и с гидроксильной ( ol ) группой). Это завершает каталитическую часть процесса, которая в основном представляет собой кислотно-основную группу. (перенос протона) реакция, в которой роль протона состоит в том, чтобы отнимать электрон от кислорода кетона.
На второй стадии енол реагирует с йодом.Изогнутые стрелки указывают на сдвиги в расположении электронов. Ниже электрон отводится с π-орбитали двойной связи одним из атомов молекулы I 2 . Это вызывает сдвиг электронов в последнем, в результате чего половина этой молекулы выбрасывается в виде иодид-иона. Другая половина йода теперь представляет собой ион йодония I + , который вытесняет протон одной из метильных групп. Образовавшийся ион карбония затем вытесняет протон -ОН с получением конечного нейтрального продукта.
Возможно, наиболее известной реакцией, катализируемой кислотой, является гидролиз (или образование) сложного эфира — реакция, с которой большинство студентов сталкивается в лабораторных курсах органической химии. Это более сложный процесс, состоящий из пяти шагов; его механизм обсуждается здесь. См. Также этот сайт У. Калгари, который описывает реакцию, катализируемую как кислотой, так и основанием.
Окислительно-восстановительный катализ
Многие реакции окисления-восстановления (переноса электрона), включая прямое окисление молекулярным кислородом, протекают довольно медленно.{2 +}} \]
Эта реакция катализируется либо Cu + , либо Cu 3+ , и скорость пропорциональна концентрации V 3+ и иона меди, но не зависит от концентрации Fe 3+ . Считается, что этот механизм состоит из двух этапов:
| 1 V 3+ + Cu 2+ V 4+ + Cu + | (нормировочная) |
| 2 Fe 3+ + Cu + Fe 2+ + Cu 2+ | (очень быстро) |
(Если в качестве катализатора используется Cu + , он сначала окисляется до Cu 2+ на этапе 2.+ \]
Катализаторы гетерогенные
Как следует из названия, гетерогенный катализатор существует как отдельная фаза (почти всегда твердая) от фазы (чаще всего газа), в которой происходит реакция. Каталитический эффект возникает из-за разрушения (часто приводящего к диссоциации) молекул реагента, вызванного их взаимодействием с поверхностью катализатора.
← Модель катализатора, состоящего из кластеров из 8-10 атомов платины (синий цвет), нанесенных на поверхность оксида алюминия.Этот катализатор эффективно удаляет атомы водорода из пропана, превращая его в промышленно важный пропилен. [источник]
Неуравновешенные силы на поверхностях
Как вы помните, одно универсальное свойство материи — это слабые силы притяжения, возникающие, когда две частицы близко подходят друг к другу. (См. Здесь для быстрого обзора.) Когда частицы имеют противоположные электрические заряды или вступают в ковалентную связь, это гораздо более сильное притяжение доминирует и определяет «химию» взаимодействующих частиц.
Молекулярные единицы в объеме твердого тела связаны со своими соседями посредством этих сил, которые действуют в противоположных направлениях, удерживая каждую из них под своего рода «натяжением», которое ограничивает ее движение и способствует связности и жесткости твердого тела.
На поверхности любого конденсированного вещества все обстоит иначе. Атомы или молекулы, которые находятся на поверхности, испытывают несбалансированные силы, которые не позволяют им принимать такие же низкие потенциальные энергии, которые характерны для внутренних единиц.(То же самое происходит с жидкостями и вызывает множество межфазных эффектов, таких как поверхностное натяжение.)
Но в случае твердого тела, в котором силы притяжения обычно сильнее, происходит нечто гораздо более значительное. Молекулярные единицы, которые находятся на поверхности, можно рассматривать как частично погруженные в нее, а их выступающие части (и возникающие из них межмолекулярные притяжения) открыты внешнему миру. Сила притягивающего силового поля, которое исходит от твердой поверхности, различается по силе в зависимости от природы атомов или молекул, составляющих твердое тело.
Не путайте сорбцию d с сорбцией b ; последнее относится к массовому поглощению вещества внутри пористого материала. На микроскопическом уровне, конечно, поглощение также связано с адсорбцией. Процесс, при котором молекулы газа или жидкости вступают в контакт с твердой поверхностью и прикрепляются к ней, известен как адсорбция , . Адсорбция почти всегда является экзотермическим процессом, и ее сила обычно выражается энтальпией или «теплотой» адсорбции Δ H адс .
Хемосорбция и физическая сорбция
Обычно различают две общие категории адсорбции, в зависимости от степени воздействия на электронную или связывающую структуру присоединенной молекулы. Когда силы притяжения возникают из-за относительно слабых ван-дер-ваальсовых взаимодействий, такой эффект невелик, и Δ H ad имеет тенденцию быть малым. Это состояние описывается как физическая адсорбция (физическая адсорбция ). Физическая адсорбция газа на поверхности энергетически похожа на конденсацию газа в жидкость, она обычно создает несколько слоев адсорбированных молекул и протекает с нулевой энергией активации.
Более важное значение для каталитических явлений имеет хемосорбция , при которой адсорбат связан с поверхностью химической связью. Возникающее в результате нарушение электронной структуры адсорбированных частиц «активирует» их и делает их подверженными химической реакции (часто диссоциации), которая не может быть легко достигнута путем термической активации в газовой или жидкой фазе. В отличие от физадсорбции, хемосорбция обычно включает энергию активации (обеспечиваемую Δ H адс ), и адсорбированные частицы всегда являются монослоем.
Механизмы реакций на поверхностях
Диссоциативная адсорбция
Простейшим гетерогенным процессом является хемосорбция с последующим разрывом связи, как описано выше. Наиболее распространенным и тщательно изученным из них является диссоциация водорода, которая происходит на поверхности большинства переходных металлов. Одиночный 1 s электрон каждого атома водорода координируется с d орбиталями металла, образуя пару хемосорбционных связей (обозначенных красными пунктирными линиями).Хотя эти новые связи более стабильны, чем простая ковалентная связь, которую они заменяют, образующиеся в результате атомы водорода могут перемещаться по поверхности благодаря непрерывной протяженности орбитальной зоны проводимости d .
Механизм Eley-Rideal
Альтернативный механизм исключает вторую стадию хемосорбции; адатомы кислорода взаимодействуют непосредственно с газообразными молекулами CO , заменяя хемосорбционную связь новой связью C – O, когда они проносятся по поверхности:
Примеры обоих механизмов известны, но механизм Ленгмюра-Хиншелвуда более важен, поскольку он использует активацию адсорбированного реагента.В случае окисления окиси углерода исследования, включающие эксперименты с молекулярными пучками, подтверждают эту схему. Ключевым свидетельством является наблюдение короткого промежутка времени между контактом молекулы CO с поверхностью и высвобождением CO 2 , что позволяет предположить, что CO остается хемосорбированным в течение этого интервала.
Принцип Сабатье
Чтобы быть эффективными, эти процессы адсорбции, реакции и десорбции должны быть организованы таким образом, чтобы в значительной степени зависеть от свойств катализатора по отношению к хемосорбционным свойствам (Δ H адс ) реагентов и продуктов.
- Адсорбция реагента на каталитической поверхности ( 2 ) должна быть достаточно сильной, чтобы нарушить связывание внутри вещества, чтобы диссоциировать или активировать его;
- Если полученные фрагменты должны мигрировать в другие места на поверхности ( 3-4 ), их хемосорбция должна быть достаточно слабой, чтобы допустить это движение, но не настолько малой, чтобы они ускользнули, прежде чем у них появится возможность среагировать;
- Продукт должен иметь достаточно малые значения Δ H ad , чтобы обеспечить их быструю десорбцию с катализатора ( 5 ), чтобы поверхность была освобождена для повторения цикла
Важность выбора катализатора, который обеспечивает надлежащий баланс теплоты адсорбции различных компонентов реакции, известна как Принцип Сабатье , но иногда его называют «правильным» или «принципом Златовласки».Помните историю Златовласки и трех медведей? … или посмотрите это видео на UTube.
В применении к катализу этот принцип часто иллюстрируется «вулканической диаграммой», на которой скорость катализированной реакции представлена как функция Δ H адс субстрата, такого как H 2 , на поверхность переходного металла.
График слева показывает относительную эффективность различных металлов в катализе разложения муравьиной кислоты HCOOH.Вертикальная ось отложена как температура. Идея состоит в том, что чем лучше катализатор, тем ниже температура, необходимая для поддержания заданной скорости.
Каталитический цикл
Этот термин относится к идеализированной последовательности стадий между адсорбцией реагента на катализаторе и десорбцией продукта, завершающейся восстановлением катализатора до его исходного состояния. Типичный каталитический цикл гидрирования пропена проиллюстрирован ниже.
Эта конкретная реакция
H 3 C – CH = CH 2 + H 2 → H 3 C – CH 2 –CH 3
происходит спонтанно только в обратном направлении, но он представляет процесс, используемый для гидрогенизации двойных углерод-углеродных связей в растительных маслах с получением твердых насыщенных жиров, таких как маргарин.
Катализатор отравления и разрушения
Отравление катализатором, вызванное необратимым связыванием вещества с его поверхностью, может быть постоянным или временным. В последнем случае катализатор можно регенерировать, обычно путем нагревания до высокой температуры. В организмах многие вещества, известные нам как «яды», действуют как каталитические яды на ферменты. Если катализаторы действительно остаются неизменными, они должны работать вечно, но на практике могут происходить различные события, ограничивающие срок службы многих катализаторов.
- Примеси в исходном сырье или продуктах побочных реакций могут постоянно связываться с достаточным количеством активных центров, чтобы со временем снижать каталитическую эффективность, «отравляя» катализатор.
- Физическое разрушение катализатора или его носителя, часто вызываемое высокими температурами, иногда используемыми в промышленных процессах, может уменьшить эффективную площадь поверхности или доступность реагентов к активным центрам.
Катализаторы, как правило, довольно дороги, поэтому желательно, чтобы их можно было повторно обрабатывать или регенерировать для восстановления их активности.Обычной промышленной практикой является периодическая остановка технологических установок для замены отработанных катализаторов.
Как работают гетерогенные катализаторы
Фактические механизмы, с помощью которых адсорбция молекулы на каталитической поверхности способствует разрыву связи, сильно варьируются от случая к случаю.
Мы приводим здесь только один пример — диссоциацию диксо гена O 2 на поверхности катализатора, способного временно отдавать электрон, который попадает на кислородную антисвязывающую молекулярную орбиталь, которая явно дестабилизирует связь O – O.(Как только связь разорвана, электрон возвращается к катализатору.)
Типы каталитически активных поверхностей
Гетерогенные катализаторы в основном зависят от одного или нескольких из следующих типов поверхностей:
Некоторые факторы, влияющие на эффективность катализатора
Поскольку гетерогенный катализ требует прямого контакта между реагентами и каталитической поверхностью, площадь активной поверхности идет вверху списка. В случае металлической пленки это не то же самое, что номинальная площадь пленки, измеренная линейкой; на микроскопическом уровне даже кажущиеся гладкими поверхности очень неровны, а некоторые полости могут быть слишком маленькими для размещения молекул реагентов.
Рассмотрим, например, что куб платины размером 1 см (стоимостью примерно 1000 долларов) имеет номинальную площадь поверхности всего 6 см 2 . Если его разбить на 10 12 меньших кубов со сторонами 10 –6 м, общая площадь поверхности составит 60 000 см 2 , что в принципе способно увеличить скорость реакции, катализируемой Pt, в раз. из 10 4 . Эти очень мелкоизмельченные (и часто очень дорогие) металлы обычно прикрепляются к инертной опорной поверхности для максимального воздействия на них реагентов.
Рельеф поверхности. На микроскопическом уровне даже внешне гладкая поверхность имеет ямки и неровности, и некоторые участки будут более активными, чем другие. Проникновение молекул в некоторые из более мелких каналов пористой поверхности и из них может стать ограничивающим скорость.
В остальном гладкая поверхность всегда будет иметь множество дефектов, таких как ступеньки и углы, которые обеспечивают большую открытость и могут быть либо единственными активными центрами на поверхности, либо чрезмерно активными, чтобы постоянно связываться с реагентом, уменьшая активную площадь поверхность.В одном исследовании было определено, что дефекты излома, составляющие всего 5 процентов поверхности платины, ответственны за более 90% каталитической активности в определенной реакции.
Стерические факторы
Когда хемосорбция происходит в двух или более местах на реагенте, для эффективного катализа необходимо, чтобы расстояние между активными центрами на каталитической поверхности было таким, чтобы поверхностные связи могли образовываться без значительного углового искажения.
Таким образом, активация двойной связи этилена на поверхности никеля происходит эффективно, потому что угол между связями C-Ni и C-C близок к тетраэдрическому значению 109.Углерод 5 °, необходимый для формирования гибридной орбиты sp 3 . Точно так же можно ожидать, что гидрирование бензола должно эффективно протекать на поверхности, на которой активные центры расположены в диапазоне от 150 до 250 пм.
Это одна из причин, по которой многие металлические катализаторы проявляют разную каталитическую активность на разных поверхностях кристаллов.
Некоторые специальные типы гетерогенных катализаторов
Металлические кластерные катализаторы
По мере уменьшения размера частиц катализатора доля более открытых ступенчатых, краевых и угловых атомов увеличивается.Крайний случай имеет место с наноразмерными (1-2 нм) металлическими кластерными структурами, обычно состоящими из 10-50 атомов.
[ссылка] →
Металлическое золото, хорошо известное своей химической инертностью, проявляет очень высокую каталитическую активность, когда оно осаждается в виде металлических кластеров на оксидном носителе. Например, O 2 легко диссоциирует на кластеры Au 55 , которые, как было обнаружено, эффективно катализируют окисление углеводородов [статья].
Цеолитные катализаторы
Цеолиты представляют собой глиноподобные твердые частицы алюмосиликата, которые образуют микропористые структуры с открытым каркасом, которые могут содержать связанные клетки, полости или каналы, размеры которых могут быть адаптированы к размерам реагентов и продуктов.Для тех молекул, которые способны диффундировать через эти пространства, цеолиты фактически являются «всей поверхностью», что делает их очень эффективными. Такая избирательность по размеру делает их важными для адсорбции, разделения, ионного обмена и каталитических применений. Многие цеолиты встречаются в виде минералов, но другие получают синтетическим путем, чтобы оптимизировать их свойства.
В качестве катализаторов цеолиты обладают рядом преимуществ, которые сделали их особенно важными в операциях «зеленой химии», в которых количество стадий обработки, количество нежелательных побочных продуктов и объемы потоков отходов сведены к минимуму.
Ферменты и биокатализ
«Кувшин вина, буханка хлеба и…» Ферменты!
Это искажение уже искаженного перевода Роберта Фитцджеральда знаменитого катрена из чудесного Рубайят Омара Хайяма подчеркивает центральную роль, которую ферменты и их технологии играли в цивилизации с древних времен.
← Иллюстрация Эдмунда Дюлака (1882-1953)
Ферментация и виноделие были частью истории и культуры человечества не менее 8000 лет, но признание роли катализа в этих процессах пришлось отложить до конца девятнадцатого века.К 1830-м годам было обнаружено множество подобных агентов, например, тех, которые способствуют перевариванию белков в желудке. Термин «фермент», означающий «дрожжевой», был придуман немецким физиологом Вильгельмом Кюне в 1876 году. В 1900 году Эдуард Бухнер (1860-1917, Нобелевская премия по химии 1907 года) показал, что брожение, которое, как ранее считалось, зависит от загадочного «Жизненная сила», содержащаяся в живых организмах, таких как дрожжи, может быть достигнута бесклеточным «прессованным соком», который он выжал из дрожжей.
К этому времени было признано, что ферменты являются формой катализатора (термин, введенный Берцелиусом в 1835 году), но их точная химическая природа оставалась под вопросом. Оказалось, что они связаны с белками, но общее осознание того, что ферменты — это белки , началось только в 1930-х годах, когда был кристаллизован первый чистый фермент, и не стало общепринятым до 1950-х годов. Теперь ясно, что почти все ферменты являются белками, за исключением небольшого, но важного класса ферментов на основе РНК, известных как рибозимы.
Белки состоят из длинных последовательностей аминокислот, связанных амидными связями; эта последовательность определяет первичную структуру белка. Их огромный размер (обычно 200-2000 аминокислотных единиц с общей молекулярной массой 20 000 — 200 000) позволяет им складываться сложными способами (известными как вторичные и третичные структуры), чьи конфигурации необходимы для их каталитической функции.
Поскольку ферменты обычно намного больше, чем молекулы реагентов, на которые они действуют (известные в биохимии как субстраты ), ферментативный катализ в некотором смысле похож на гетерогенный катализ.Основное отличие состоит в том, что связывание субстрата с ферментом происходит гораздо более избирательно.
Прекурсоры и кофакторы
Большинство ферментов возникают в виде неактивных предшественников ( зимогенов ), которые превращаются в свои активные формы в то время и в том месте, где они необходимы.
- Например, ферменты, которые приводят к свертыванию крови, должны оставаться неактивными до фактического начала кровотечения; основным активирующим фактором является воздействие на кровь белков поврежденной стенки сосуда.
- Фермент, который катализирует образование лактозы (молочного сахара) в молочной железе, образуется во время беременности, но остается неактивным до момента рождения, когда гормональные изменения заставляют элемент-модификатор связываться с ферментом и активировать его.
Превращение в активную форму может включать простое разрушение белка путем гидролиза соответствующей пептидной связи или присоединения фосфатной или аналогичной группы к одному из аминокислотных остатков.
Многие ферментные белки также требуют «вспомогательных» молекул, известных как кофакторов , чтобы сделать их каталитически активными.Это могут быть простые ионы металлов (многие из микропитательных ионов Cu, Mn, Mo, V и т. Д.) Или более сложные органические молекулы, которые называются коферментами . Многие из последних — это то, что мы обычно называем витаминов . Другие молекулы, известные как ингибиторы , снижают активность ферментов; Так действуют многие лекарства и яды.
Фермент-субстратный комплекс
Стандартная модель кинетики фермента состоит из двухэтапного процесса, в котором фермент обратимо связывается со своим субстратом S (реагентом) с образованием комплекса фермент-субстрат ES:
Комплекс фермент-субстрат играет роль, аналогичную роли активированного комплекса в традиционной кинетике, но основная функция фермента — стабилизировать переходное состояние.
На второй, по существу необратимой стадии, продукт и фермент высвобождаются:
Общая кинетика процесса была разработана в 1913 году немецким биохимиком Леонором Михаэлисом и его канадской ученицей Мод Ментен.
Основная кинетическая трактовка этого процесса включает предположение, что концентрации [E] и [ES] достигают стационарных значений, которые не меняются со временем. (Подробное лечение, выходящее за рамки этого курса, можно найти здесь.)
Общий процесс описывается уравнением Михаэлиса-Ментен , которое изображено здесь. Константа Михаэлиса k M определяется, как показано, но может быть упрощена до константы диссоциации ES k -1 / k 1 в случаях, когда диссоциация комплекса является скоростной. предельный шаг. Величина v max не наблюдается напрямую, но может быть определена из k M , как показано здесь.
Ферменты белки
Чтобы понять ферменты и то, как они катализируют реакции, сначала необходимо рассмотреть несколько основных понятий, касающихся белков и аминокислот, из которых они состоят.
Аминокислоты: полярные, неполярные, положительные, отрицательные.
21 аминокислота, из которых состоят белки, обладают основной структурой, показанной здесь, где R представляет собой водород или боковую цепь, которая сама может содержать дополнительные группы –NH 2 или –COOH.Оба типа групп могут образовывать водородные связи с водой и с полярными частями субстратов и, следовательно, вносят свой вклад в полярность и гидрофильность аминокислот. Боковые цепи, которые содержат более длинные углеродные цепи и особенно бензольные кольца, имеют противоположный эффект и имеют тенденцию делать аминокислоту неполярной и гидрофобной.
pK a ионизируемой группы — это pH, при котором половина этих групп будет ионизирована. Например, если группа –COOH имеет pK , то это , равное 2.5, он будет почти полностью существовать в этой форме при pH около 1,5 или меньше, и в виде отрицательного иона –COO — при pH выше около 3,5.
Обе группы –NH 2 и –COOH ионизируются (т.е. они могут действовать как доноры или акцепторы протонов), и когда они находятся в ионной форме, они будут иметь электрический заряд. Группы –COOH имеют pKa в диапазоне 1,8–2,8 и, следовательно, будут находиться в их ионизированных формах –COO – при обычных значениях клеточного pH около 7.4. РКа аминогруппы составляет около 8,8-10,6, поэтому они также обычно находятся в ионизированных формах NH 3 + .
Это означает, что при обычном клеточном pH как карбоксильная, так и аминогруппа будут ионизированы. Но поскольку заряды имеют противоположные знаки, аминокислота, не имеющая дополнительных ионизируемых групп в боковой цепи, будет иметь нулевой чистый заряд. Но если боковая цепь содержит дополнительную амино- или карбоксильную группу, аминокислота может нести чистый электрический заряд.На следующей диаграмме показаны типичные аминокислоты, которые попадают в каждую из описанных выше групп.
Для ясности здесь показаны только заряды в боковых цепях, но, конечно, карбоксильные и аминогруппы в головном конце каждой аминокислоты также будут ионизированы. Маленькие зеленые числа — это значения pK и . См. Здесь полный список аминокислот, которые встречаются в белках.
Белки
Белки состоят из одной или нескольких цепочек аминокислот, связанных друг с другом пептидными связями путем удаления молекулы воды.
Продукт, показанный выше, называется пептидом , в частности, это дипептид , потому что он содержит две аминокислот остатков (то, что осталось после удаления воды). Белки — это просто очень длинные полипептидные цепи, или их комбинации. (Различие между длинным полипептидом и маленьким белком довольно нечеткое!)
Глобулярные белки
Большинство ферментов попадают в категорию глобулярных белков .В отличие от волокнистых белков , которые образуют структурные компоненты тканей, глобулярные белки растворимы в воде и редко имеют какие-либо систематические третичные структуры. Они состоят из одной или нескольких аминокислотных цепочек (« пептид »), которые складываются в различные формы, которые можно примерно описать как сферические — отсюда термин «глобулярный» и суффикс «глобин», который часто добавляется к их названия, как в «гемоглобин».
Сворачивание белка — это спонтанный процесс, на который влияет ряд факторов.Одна из них, очевидно, является их первичной аминокислотной последовательностью, которая способствует образованию внутримолекулярных связей между аминокислотами в различных частях цепи. Они состоят в основном из водородных связей, хотя нередки дисульфидные связи S-S между серосодержащими аминокислотами.
Помимо этих внутримолекулярных сил, важную роль играют взаимодействия с окружающей средой. Наиболее важным из них является гидрофобный эффект , который способствует конформациям складчатости, в которых полярные аминокислоты (которые образуют водородные связи с водой) находятся снаружи, в то время как так называемые гидрофобные аминокислоты остаются в защищенных местах в пределах складки.
Как работают ферменты: активный центр
Каталитический процесс, опосредованный ферментом, происходит в углублении или щели, в результате чего субстрат подвергается воздействию лишь нескольких из сотен и тысяч аминокислотных остатков в белковой цепи. Высокая специфичность и активность ферментативного катализа чувствительно зависят от формы этой полости и от свойств окружающих аминокислот
В 1894 году, задолго до того, как стало ясно, что ферменты являются белками, немецкий химик Эмиль Фишер предложил так называемую модель «замок-и-ключ» как способ понять, как данный фермент может специфически действовать только на один вид молекул субстрата.Эта модель, по сути, является развитием той, которую мы все еще используем для объяснения гетерогенного катализа.
Хотя базовая модель с замком и ключом продолжает оставаться полезной, она была изменена в то, что теперь называется моделью с принудительной подгонкой, . Это предполагает, что, когда субстрат входит в активный сайт и взаимодействует с окружающими частями аминокислотной цепи, он изменяет форму активного сайта (и, возможно, других частей фермента), чтобы он мог более полно взаимодействовать с субстратом.
Одним из важных шагов в этом процессе является выдавливание любых молекул воды, которые связаны с субстратом и могут помешать его оптимальному расположению.
В активном центре специфические взаимодействия между субстратом и соответствующим образом заряженными, гидрофильными и гидрофобными аминокислотами активного центра затем стабилизируют переходное состояние, искажая молекулу субстрата таким образом, чтобы привести к переходному состоянию, имеющему существенно более низкую активацию. энергии, чем может быть получен обычным неферментативным катализом.За пределами этой точки основные каталитические стадии довольно обычны, причем наиболее распространенными являются кислотно-щелочной и нуклеофильный катализ.
Для очень ясной и поучительной иллюстрации типичной многоступенчатой последовательности типичной реакции, катализируемой ферментами, см. Эту страницу онлайн-учебника Марка Бишопа, из которого взята эта иллюстрация.
Регулирование и ингибирование ферментов
Если бы все ферменты в организме были активны все время, результатом был бы хаос.Большинство клеточных процессов, таких как производство и использование энергии, деление клеток и расщепление продуктов метаболизма, должны протекать в изящно отлаженной и точно настроенной манере, как в большом симфоническом оркестре; Здесь нет места джаз-импровизации!
Природа придумала различные способы достижения этого; мы описали действие предшественников и коферментов. Здесь мы сосредоточимся на одном из наиболее важных (и химически интересных) регуляторных механизмов.
Аллостерическая регуляция: настройка активного сайта
Существует важный класс ферментов, которые обладают особыми сайтами (отличными от каталитически активных сайтов), с которыми определенные внешние молекулы могут обратимо связываться.Хотя эти аллостерических сайтов , как их называют, могут быть довольно далеко от каталитических сайтов, эффект связывания или высвобождения этих молекул должен запускать быстрое изменение в паттерне укладки фермента, которое изменяет форму фермента. активный сайт. Эффект состоит в том, чтобы позволить сигнальной или регуляторной молекуле (часто очень маленькой, такой как NO) модулировать каталитическую активность активного центра, эффективно включая или выключая фермент.
В некоторых случаях продукт катализируемой ферментом реакции может сам связываться с аллостерическим сайтом, снижая активность фермента и, таким образом, обеспечивая отрицательную обратную связь, которая помогает поддерживать продукт в желаемой концентрации.Считается, что таким образом регулируются концентрации ионов в плазме, таких как кальций, и энергоснабжающего АТФ.
Аллостерические ферменты — это больше, чем просто катализаторы: они действуют как контрольные точки в метаболических и клеточных сигнальных сетях. Аллостерические ферменты часто стоят в начале последовательности ферментов в метаболической цепи или в точках разветвления, где две такие цепи расходятся, действуя очень сильно. как светофоры на перегруженных перекрестках.
Ингибирование ферментов
Как и в случае с гетерогенными катализаторами, некоторые молекулы, отличные от нормального субстрата, могут быть способны проникать и удерживаться в активном центре, чтобы конкурентно ингибировать активность фермента.Так действуют пенициллин и родственные ему антибиотики; эти молекулы ковалентно связываются с аминокислотными остатками в активном центре фермента, который катализирует образование важного компонента стенок бактериальных клеток. Когда клетка делится, новообразованное потомство по существу распадается.
Ферменты вне клетки
Концепция художника Майка Перкинса об иммобилизованном ферменте внутри функционализированной нанопористой поры кремнезема. Фермент показан зеленым цветом, а положительно заряженные области показаны красным.Синие структуры внутри пор представляют собой отрицательно заряженные функциональные группы, добавленные к мезопористому диоксиду кремния, чтобы создать благоприятную химическую среду для фермента. Благоприятная среда в каждой поре привлекает молекулу фермента, чтобы двигаться в незанятую пору. Эта среда стабилизирует и увеличивает химическую активность фермента, позволяя ему преобразовывать вредные материалы субстрата (фиолетовые частицы) в полезные или безвредные продукты (желтые и красные частицы). [ссылка на сайт]Ферменты уже очень давно широко используются в пищевой, целлюлозно-бумажной и детергентной промышленности, но в основном в виде неочищенных экстрактов целых клеток.
В последние годы развитие биотехнологии и постепенный переход промышленности от нефтяного сырья и растворителей к так называемой «зеленой» химии сделали ферменты более привлекательными в качестве промышленных катализаторов. По сравнению с последними очищенные ферменты имеют тенденцию быть дорогими, трудно перерабатываемыми и нестабильными за пределами довольно узких диапазонов температуры, pH и состава растворителя.
Ферменты иммобилизованные
Многие проблемы, связанные с использованием свободных ферментов, можно преодолеть путем иммобилизации фермента.Это можно сделать несколькими способами:
- Связывание фермента с инертным твердым поддерживающим материалом
- Впервые это было сделано в 1916 году с использованием древесного угля или гидроксида алюминия. С 1960-х годов, когда этот метод стал более популярным, широкое распространение получили синтетические полимеры, часто предназначенные для конкретного применения. Также были использованы некоторые природные биополимеры, такие как целлюлоза или крахмал, и неорганические твердые вещества, такие как диоксид кремния и оксид алюминия.
- Улавливание фермента в пористом материале
- Полимеризация акриламида в присутствии фермента дает пористый гель, который обеспечивает свободную диффузию субстрата и продуктов. Другие материалы, используемые для этой цели, включают натуральные гели, такие как альгинат (полученный из морских водорослей), пористый кремнезем или твердые частицы керамики и цеолиты.
- Сшитые ферменты
- Если фермент химически связан с другим веществом (обычно очень маленькой молекулой), образующееся твердое вещество, известное как сшитый агрегат фермента, может сохранять высокую каталитическую эффективность в очень широком диапазоне условий. Более недавняя связанная разработка включает осаждение менее очищенной формы фермента (например, путем добавления этиленгликоля или сульфата аммония) с последующим сшиванием полученных агрегатов.
Ссылки: больше для изучения
Катализ
- Гади Ротенберг- Катализ: концепции и экологические приложения (Wiley-VCH, 2008)
- R.A. ван Сантан, М. Нейрок — Молекулярный гетерогенный катализ: концептуальный и вычислительный подход (Wiley-VCH, 2006)
- История катализа: список веб-страниц, включая пятьдесят лет катализа — список основных достижений в области 1949–1999. Более подробные страницы истории из UK
- Катализ и его приложения — очень читаемый и интересный отчет о приложениях катализа в различных промышленно важных областях.
Авторы и авторство
Роль катализатора — Институт творческих сообществ Рыцаря
Примите участие
Что такое Community Catalyst?
Катализаторы сообщества KCCI — это агенты изменений. Они берут на себя волонтерские обязательства по обучению, вовлечению и предоставлению согражданам возможности построить более аутентичное, устойчивое и процветающее сообщество. Нажмите кнопку ниже, чтобы узнать больше о предстоящем проекте, а также присоединитесь к нашему списку рассылки, чтобы следить за нашей деятельностью.
Подробнее о текущем проекте
Каждый катализатор будет:
- Развитие партнерских отношений и взаимоотношений для реализации проекта чувства места в течение года
- Примите участие в двухдневном тренинге, на котором они изучат концепции творческого сообщества, местную экономику и глобальные тенденции экономического развития, а также узнают больше о проекте
- Узнайте о стратегиях местного сообщества и экономических принципах, связанных с размещением
- Будь командным игроком
- Будьте провидцами
- Посвящать в среднем 2–3 часа в неделю в течение 12 месяцев
Обязанности и обязанности включают:
- Подготовка к году
- Изучение чтений и текущих практик
- Обсудите с другими членами сообщества регион, его сильные стороны, ресурсы и потребности
- Проанализировать последние экономические данные
- Полноценное участие в командном обучении и семинарах по выработке коллективного видения
- Развивайте инициативу
- Посещать собрания команды
- Будьте активным и конструктивным членом команды
- Координировать усилия
- Поддерживайте простой поток информации с другими катализаторами и инициативами
- Посещать собрания команды КТПП
- Связать все общественные коммуникации с KCCI в целом
- Регулярно встречаться с KCCI
- Быть готовым внести свой вклад в успех KCCI гибкими способами
- Заинтересуйте свое сообщество
- Практикуйте и продвигайте принципы KCCI и Творческого сообщества и используйте их в своей инициативной работе
- Поделитесь своим анализом и пониманием местной и региональной экономики
- Будьте выразителем видения и инициатив Catalysts
- Использование доступных ресурсов и доступ к ним
- Принять и применить на практике концепцию, согласно которой задача каждого Catalyst — обучать, учиться и действовать в соответствии с их инициативой и видением KCCI
- Развивайте и поддерживайте инклюзивный процесс:
- Сопротивляйтесь продвижению повестки дня только избранных: процесс KCCI не основан на особых интересах отдельных лиц
- Продвигайте видение и интересы KCCI и вашей инициативы через участие сообщества: постоянно приглашайте и вербуйте людей, которые не являются оригинальными Катализаторами, для участия в вашей инициативе
- Достижение консенсуса и согласованных действий
- Продемонстрируйте, что Катализаторы являются руководителями и активистами сообщества, которое привлекает творческих, вовлеченных, успешных, продуктивных и эффективных людей
- Структурируйте свою инициативу таким образом, чтобы обеспечить разнообразие и широкое участие
- Укажите, как всеобъемлющая миссия и цели вашей инициативы могут поддержать и поощрить все ваши усилия к концу года KCCI:
- Если возможно, найдите способы освободить место для дополнительных действий, которые были предложены вам сообществом, когда вы работаете над своей инициативой
- Будьте открыты для мысли о том, что некоторые части вашей инициативы или группы могут захотеть или нуждаться в самостоятельной деятельности, которая получит вашу поддержку и поддержку
- Найдите и разработайте методы для 1) создания беспроигрышных ситуаций и 2) сотрудничества и взаимодействия с более широким сообществом
- Разработайте планы, обеспечивающие непрерывность усилий вашей инициативы по прошествии двенадцати месяцев: ваше наследие должно открывать двери для других, чтобы они помогали продвигать усилия вашей инициативы
- Дайте себе достаточно времени, чтобы установить конструкции, чтобы поддержать ваши усилия и надстроить их
Аутентификация — роли | Онлайн-справка
Роли определяют набор разрешений и уровень доступа, предоставляемый конечному пользователю для доступа или изменения компонентов вашего приложения.Вы можете определить области и права доступа для определенных компонентов Catalyst для каждой роли и назначить роль каждому пользователю. Эта категоризация позволяет пользователям вашего приложения получать доступ только к тем функциям, которые им важны.
Роли в настоящее время применяются к хранилищу данных и хранилищу файлов. Вы можете определить области и разрешения каждой таблицы для каждой роли пользователя в хранилище данных. Эти области и разрешения также применимы к поиску и ZCQL по расширению, поскольку эти компоненты тесно связаны с хранилищем данных.Точно так же вы можете определить права доступа к папке для каждой роли в хранилище файлов.
По умолчанию создаются две роли пользователей:
- Администратор приложения: Пользователи, которым назначена роль администратора приложения, по сути, по умолчанию имеют административный доступ к приложению. Вы можете переопределить это и определить их уровни доступа к таблицам в хранилище данных и к папкам в хранилище файлов.
- Пользователь приложения: Пользователи, которым назначена роль пользователя приложения, по умолчанию имеют доступ к приложению на уровне конечного пользователя.Вы также можете переопределить это и определить их уровни доступа в хранилище данных и хранилище файлов.
В дополнение к этим ролям вы можете создавать свои собственные пользовательские роли и определять их разрешения.
Catalyst позволяет установить роль пользователя в качестве роли по умолчанию . Когда роль пользователя задана как роль по умолчанию, после этого пользователи, добавленные в ваше приложение, автоматически назначаются этой роли, и им становятся доступны разрешения, которые были установлены для этой роли.
Преимущества
- Роли пользователей позволяют группировать пользователей приложения на основе уровней доступа и предоставленных им разрешений.
- Роли помогают предотвратить несанкционированный доступ для просмотра или изменения данных приложения, а также повысить безопасность ресурсов и данных в вашем приложении.
- Вы можете настроить области и предоставить определенные разрешения для таких действий, как просмотр данных таблицы, удаление строк из таблицы или загрузка файлов в определенные папки в хранилище данных и хранилище файлов для пользовательских ролей.
- Они также позволяют вам контролировать поток данных и дизайн вашего приложения в соответствии с вашими потребностями. Вы можете создать свое приложение, помня о многоуровневом доступе, который вы можете предоставить различным группам пользователей.
Создание новой роли
Чтобы создать новую роль в Catalyst, необходимо клонировать существующую роль. Это клонирует набор разрешений от родительской роли к новой роли.
Примечание: В настоящее время вы не сможете удалять роли из Catalyst.Положение об удалении ролей будет выпущено в ближайшее время.
Чтобы создать новую роль пользователя для приложения Catalyst:
- Щелкните вкладку Roles в Authentication .
- Щелкните Добавить роли в разделе ролей .
- Введите имя роли во всплывающем окне.
- Выберите существующую роль для клонирования разрешений в поле Clone Role .
- Введите описание роли.
- Включите IsDefault , чтобы сделать эту роль ролью по умолчанию в вашем приложении Catalyst.
- Щелкните Create .
Роль теперь будет отображаться в разделе Роли вместе с подробностями. Для новой роли будет создан уникальный идентификатор роли, который можно использовать для ссылки на роль при добавлении пользователя из API или SDK. Роли администратора приложения и пользователя приложения уже будут иметь идентификаторы ролей по умолчанию.
Вы можете включить или отключить роль в качестве роли по умолчанию с помощью переключателя.
Переименовать роль
Чтобы переименовать роль пользователя:
- Щелкните значок многоточия для роли, которую нужно переименовать, и щелкните Изменить в разделе Роли .
- Введите новое имя роли и нажмите Введите .
Влияние катализаторов на скорость реакции
Катализаторы и энергия активации
Для увеличения скорости реакции нужно увеличить количество успешных столкновений.Один из возможных способов сделать это — предоставить альтернативный способ протекания реакции, который имеет более низкую энергию активации.
Другими словами, переместить энергию активации на график так:
Как и раньше, частицы, которым не хватает энергии в определенное время, когда-нибудь в будущем будут получать энергию от случайных столкновений, так же как другие частицы будут терять энергию. Вы не должны представить себе, что частицы в синей области графика никогда не могут реагировать — учитывая время, они будут.
Добавление катализатора дает именно такой эффект сдвига энергии активации. Катализатор обеспечивает альтернативный путь реакции. Этот альтернативный путь имеет более низкую энергию активации. Отображается в энергетическом профиле:
Предупреждение!
Будьте очень осторожны, если вас спросят об этом на экзамене. Правильная форма слова —
«Катализатор обеспечивает альтернативный путь реакции с более низкой энергией активации.»
Это не , а не «снижает энергию активации реакции». Там есть тонкое различие между двумя утверждениями, которое легко проиллюстрировать простой аналогией.
Предположим, у вас есть гора между двумя долинами, так что единственный способ добраться из одной долины в другую — через гору. Из одной долины в другую удастся попасть только самым активным людям.
Теперь предположим, что через гору прорезан туннель.Гораздо большему количеству людей теперь удастся добраться из одной долины в другую по этому более легкому маршруту. Можно сказать, что туннельный маршрут имеет более низкую энергию активации, чем переход через гору.